- Understanding Alzheimer's Disease
Understanding Alzheimer's Disease
What is Alzheimer's disease?
Alzheimer's disease is a brain disease that slowly destroys memory and thinking skills. It is a progressive disease, which means it gets worse over time.
Alzheimer's disease is irreversible. People with Alzheimer's eventually lose the ability to carry out the simplest of tasks.
Alzheimer's is the most common cause of dementia among people aged 65 and older. Dementia is loss of the ability to think and remember things that is severe enough that a person has trouble doing day-to-day activities.
About 5.8 million people in the United States have Alzheimer's disease.
Rarely, people younger than 65 can have Alzheimer's. This is called early-onset Alzheimer's disease . Around one of every 20 people with Alzheimer's have early-onset Alzheimer's disease.
You're more likely to get Alzheimer's if one or more of your close family members – parents, brothers, or sisters – has it.
Common early symptoms of Alzheimer's disease
The most common early symptom of Alzheimer's disease is trouble remembering recent events.
Other early symptoms may include
- Having trouble completing daily tasks, and
- Getting lost when driving on a route the person used to know well
These symptoms are not the same as normal forgetfulness, which can happen to all of us as we get older. For example, if you sometimes forget about an appointment, forget a person's name, or misplace your keys, it doesn't mean you have Alzheimer's.
Symptoms of mild Alzheimer's disease
A person with mild Alzheimer's disease may
- Need help with usual tasks (like managing finances, planning meals, and keeping appointments)
- Have trouble sleeping, and
- Become anxious or depressed.
Symptoms of moderate Alzheimer's disease – When Alzheimer's disease worsens
As symptoms of Alzheimer's disease get worse, a person may
- Have some trouble recognizing family members and friends.
- Need help with daily activities like getting dressed.
- Become fearful or suspicious of other people.
- Believe they are seeing or hearing things that aren't real.
Symptoms of severe Alzheimer's disease – When Alzheimer's disease becomes severe
When Alzheimer's disease becomes severe , a person loses much of their ability to communicate and needs full-time help to take care of themselves.
What causes Alzheimer's disease?
Doctors don't fully understand what causes Alzheimer's disease in most people.
Most people with Alzheimer's disease are older, but just getting older doesn't cause the disease. Many people live well into their 90s without getting Alzheimer's disease.
What causes Alzheimer's disease? – Genes associated with Alzheimer's disease
Carrying a variant of a gene known as APOE increases a person's risk for Alzheimer's disease, especially the late-onset form of the disease. But not everyone who has this gene gets the disease, and people who don't have the gene can still get the disease.
Early-onset Alzheimer's disease can also be caused by an inherited change in one of three other genes, yet these genes are very uncommon.
What causes Alzheimer's disease? – Conditions that may increase risk for Alzheimer's disease
Other conditions that may increase risk for Alzheimer's disease include
- High blood pressure
- An unhealthy diet
What happens in the brain in Alzheimer's disease?
Doctors now know that changes begin happening in the brain 10-20 years or more before a person with Alzheimer's disease shows any symptoms.
Neurons are brain cells that send and receive signals to and from the brain. Everything we do as living beings – walking, thinking, forming memories – happens because of these signals. A healthy adult brain contains about 100 billion neurons.
When a person has Alzheimer's disease, tiny pieces of a protein called beta amyloid build up in between neurons, forming clusters, or plaques .
Another protein, called tau , builds up inside neurons, forming dense, thread-like tangles .
Together, these plaques and tangles block neurons from sending and receiving signals .
Because of these and other abnormal changes in the brain, neurons start to die. The first place this happens is in the parts of the brain where memories are formed.
As more neurons die, the brain starts to shrink. As Alzheimer's gets worse over time, the brain may shrink to about a third of its normal size.
What is mixed dementia?
Mixed dementia is dementia that's caused by brain changes due to Alzheimer's disease and one or more other brain diseases.
For example, a person may have dementia that's caused by both Alzheimer's disease and Parkinson's disease.
What is vascular dementia?
A person may also have both Alzheimer's disease and vascular dementia . This type of dementia occurs when the brain gets less blood than it needs. This can happen after a person has had a series of small strokes.
Can Alzheimer's disease be prevented?
Studies show there are steps you can take to reduce your risk for Alzheimer's disease.
- Don't smoke.
- Keep your body and your mind active.
- Stay in touch with family and friends.
- Eat a healthy diet.
- Maintain a healthy weight.
- Control high blood pressure.
- Drink alcohol in moderation.
- Drink coffee in moderation.
Recent studies suggest that getting vaccinated against pneumonia and the flu can also help lower your risk for Alzheimer's disease.
- National Institute on Aging. Alzheimer's Disease Fact Sheet. https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet. Reviewed 5/22/2019.
- National Institute of Neurological Disorders and Stroke. Alzheimer's Disease Information Page. Definition. https://www.ninds.nih.gov/Disorders/All-Disorders/Alzheimers-Disease-Information-Page. Last modified 3/27/2019.
- Alzheimer's Association. 2020 Alzheimer's Disease Facts and Figures. https://www.alz.org/media/Documents/alzheimers-facts-and-figures.pdf.
- Zhu XC, Tan L, Wang HF, et al. Rate of early onset Alzheimer's disease: a systematic review and meta-analysis. Ann Transl Med 2015;3:38.
- National Institute on Aging. Alzheimer's and Hallucinations, Delusions, and Paranoia. https://www.nia.nih.gov/health/alzheimers-and-hallucinations-delusions-and-paranoia. Reviewed 5/17/2017.
- National Institute on Aging. Alzheimer's Disease Genetics Fact Sheet. https://www.nia.nih.gov/health/alzheimers-disease-genetics-fact-sheet. Reviewed 12/24/2019.
- Dementia Care Central. Normal Brain vs. Alzheimer's. https://www.dementiacarecentral.com/video/video-brain-changes/.
- National Institute on Aging. What Do We Know About Diet and Prevention of Alzheimer's Disease? https://www.nia.nih.gov/health/what-do-we-know-about-diet-and-prevention-alzheimers-disease. Reviewed 11/27/2019.
- Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113):2673-2734. doi:10.1016/S0140-6736(17)31363-6
- The SPRINT MIND Investigators for the SPRINT Research Group. Effect of Intensive vs Standard Blood Pressure Control on Probable Dementia: A Randomized Clinical Trial. JAMA. 2019;321(6):553–561. doi:10.1001/jama.2018.21442
- Neafsey EJ, Collins MA. Moderate alcohol consumption and cognitive risk. Neuropsychiatr Dis Treat. 2011;7:465-484. doi:10.2147/NDT.S23159
- Driscoll I, Shumaker SA, Snively BM, et al. Relationships Between Caffeine Intake and Risk for Probable Dementia or Global Cognitive Impairment: The Women's Health Initiative Memory Study. J Gerontol A Biol Sci Med Sci. 2016;71(12):1596-1602. doi:10.1093/gerona/glw078
- Alzheimer's Association. Flu, Pneumonia Vaccinations Tied to Lower Risk of Alzheimer's Dementia [press release]. 2020 July 27. https://www.alz.org/aaic/releases_2020/vaccines-dementia-risk.asp

Slide Show - Understanding Alzheimer's Disease
- Share with family and friends:
Click here to take our SURVEY Your feedback is important to us! We will use your feedback to develop future areas of content about Alzheimer's disease which will help other patients, caregivers and families.

Other Modules:

Managing and Treating Alzheimer's Disease
Jointly provided by the Annenberg Center for Health Sciences at Eisenhower and Prime Medic Inc., in collaboration with Postgraduate Institute for Medicine.
This activity is supported by an independent educational grant from Biogen Idec.
This website is part of the Animated Patient ™ series developed by Prime Medic Inc., to provide highly visual formats of learning for patients to improve their understanding, make informed decisions, and partner with their healthcare professionals for optimal outcomes.
- Terms of Use
- Technical Requirements
- © Prime Medic Inc.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
Comprehensive Review on Alzheimer’s Disease: Causes and Treatment
Alzheimer’s disease (AD) is a disorder that causes degeneration of the cells in the brain and it is the main cause of dementia, which is characterized by a decline in thinking and independence in personal daily activities. AD is considered a multifactorial disease: two main hypotheses were proposed as a cause for AD, cholinergic and amyloid hypotheses. Additionally, several risk factors such as increasing age, genetic factors, head injuries, vascular diseases, infections, and environmental factors play a role in the disease. Currently, there are only two classes of approved drugs to treat AD, including inhibitors to cholinesterase enzyme and antagonists to N -methyl d -aspartate (NMDA), which are effective only in treating the symptoms of AD, but do not cure or prevent the disease. Nowadays, the research is focusing on understanding AD pathology by targeting several mechanisms, such as abnormal tau protein metabolism, β-amyloid, inflammatory response, and cholinergic and free radical damage, aiming to develop successful treatments that are capable of stopping or modifying the course of AD. This review discusses currently available drugs and future theories for the development of new therapies for AD, such as disease-modifying therapeutics (DMT), chaperones, and natural compounds.
1. Introduction
Alzheimer’s disease (AD) (named after the German psychiatric Alois Alzheimer) is the most common type of dementia and can be defined as a slowly progressive neurodegenerative disease characterized by neuritic plaques and neurofibrillary tangles ( Figure 1 ) as a result of amyloid-beta peptide’s (Aβ) accumulation in the most affected area of the brain, the medial temporal lobe and neocortical structures [ 1 ]. Alois Alzheimer noticed a presence of amyloid plaques and a massive loss of neurons while examining the brain of his first patient that suffered from memory loss and change of personality before dying and described the condition as a serious disease of the cerebral cortex. Emil Kraepelin named this medical condition Alzheimer’s disease for the first time in his 8th edition psychiatry handbook [ 2 , 3 ]. Progressive loss of cognitive functions can be caused by cerebral disorder like Alzheimer’s disease (AD) or other factors such as intoxications, infections, abnormality in the pulmonary and circulatory systems, which causes a reduction in the oxygen supply to the brain, nutritional deficiency, vitamin B12 deficiency, tumors, and others [ 4 , 5 ].

The physiological structure of the brain and neurons in ( a ) healthy brain and ( b ) Alzheimer’s disease (AD) brain.
At present, there are around 50 million AD patients worldwide and this number is projected to double every 5 years and will increase to reach 152 million by 2050. AD burden affects individuals, their families, and the economy, with estimated global costs of US$1 trillion annually. At present, there is no cure for Alzheimer’s disease, although there are available treatments that just improve the symptoms [ 6 , 7 ]. The purpose of this review is to give a brief description about AD diagnosis, pathology, causes, and current treatments, and to highlight the recent development of compounds that could prevent or treat AD by targeting several pathogenic mechanisms, such as Aβ and tau aggregation, and misfolding, inflammation, oxidative damage, and others.
2. Alzheimer’s Disease Diagnostic Criteria
A patient suspected to have AD should undergo several tests, including neurological examination, magnetic resonance imaging (MRI) for neurons, laboratory examinations such as vitamin B12, and other tests besides the medical and family history of the patients [ 8 ]. Vitamin (vit.) B12 deficiency has been long known for its association with neurologic problems and increasing risks of AD, according to some studies. A special marker of vit. B12 deficiency is elevated homocysteine levels, which can cause brain damage by oxidative stress, increasing calcium influx and apoptosis. Diagnoses of vit. B12 deficiency can be done by measuring serum vit. B12 level alongside complete blood count and serum homocysteine levels tests [ 9 , 10 ].
In 1984, The National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders Association (ADRDA) formed a work group (NINCDS-ADRDA) to establish a clinical diagnostic’s criteria for Alzheimer’s disease. This criteria includes: (1) probable Alzheimer’s disease, which can be diagnosed by dementia that is confirmed by neuropsychological tests, progressive memory loss, impaired daily-life activity, and other symptoms like aphasia (impairment of a language), apraxia (a motor skills disorder), and agnosia (a loss of perception). All of these symptoms can start from age 40–90, with the absence of any systemic or brain diseases, (2) possible Alzheimer’s disease can be applied in the absence of neurologic, psychiatric disorders, and the presence of another illness like systemic or brain disorder, but they are not the primary cause of dementia, and (3) definite Alzheimer’s disease, that is confirmed by histopathologic confirmation obtained from a biopsy or autopsy [ 11 , 12 ].
In 2011, The National Institute on Aging—Alzheimer’s Association made several changes and updated the 1984 NINCDS-ADRDA criteria for higher specificity and sensitivity in the diagnosis of Alzheimer’s disease. The newly proposed criteria include probable and possible AD dementia for the use in clinical settings and probable or possible AD dementia with pathophysiological evidence for research purposes, in addition to clinical biomarkers. There are two categories of Alzheimer’s disease biomarkers: (a) markers of brain amyloid such as positron emission tomography (PET) and cerebrospinal fluid (CSF), and (b) markers of neuronal injury like cerebrospinal fluid tau, fluorodeoxyglucose (FDG) for metabolic activity, and magnetic resonance imaging (MRI) for atrophy measurement [ 13 , 14 , 15 ].
3. Alzheimer’s Disease’s Neuropathology
There are two types of neuropathological changes in AD which provide evidence about disease progress and symptoms and include: (1) positive lesions (due to accumulation), which are characterized by the accumulation of neurofibrillary tangles, amyloid plaques, dystrophic neurites, neuropil threads, and other deposits found in the brains of AD patients. In addition to (2) negative lesions (due to losses), that are characterized by large atrophy due to a neural, neuropil, and synaptic loss. Besides, other factors can cause neurodegeneration such as neuroinflammation, oxidative stress, and injury of cholinergic neurons [ 16 , 17 , 18 ].
3.1. Senile Plaques (SP)
The senile plaques are extracellular deposits of beta-amyloid protein (Aβ) with different morphological forms, including neuritic, diffuse, dense-cored, or classic and compact type plaques. Proteolytic cleavage enzymes such as β-secretase and γ-secretase are responsible for the biosynthesis of Aβ deposits from the transmembrane amyloid precursor protein (APP) [ 19 , 20 , 21 ]. These enzymes cleave APP into several amino acid fragments: 43, 45, 46, 48, 49, and 51 amino acids, which reach the final forms Aβ40 and Aβ42. There are several types of Aβ monomers, including large and insoluble amyloid fibrils which can accumulate to form amyloid plaques and soluble oligomers that can spread throughout the brain. Aβ plays a major role in neurotoxicity and neural function, therefore, accumulation of denser plaques in the hippocampus, amygdala, and cerebral cortex can cause stimulation of astrocytes and microglia, damage to axons, dendrites, and loss of synapses, in addition to cognitive impairments [ 21 , 22 , 23 ].
3.2. Neurofibrillary Tangles (NFTs)
NFT are abnormal filaments of the hyperphosphorylated tau protein that in some stages can be twisted around each other to form paired helical filament (PHF) and accumulate in neuralperikaryal cytoplasm, axons, and dendrites, which cause a loss of cytoskeletal microtubules and tubulin-associated proteins. The hyperphosphorylated tau protein is the major constituent of NFTs in the brains of AD patients, and its evolution can reflect NFTs morphological stages, which include: (1) pre-tangle phase, one type of NFT, where phosphorylated tau proteins are accumulated in the somatodendritic compartment without the formation of PHF, (2) mature NFTs, which are characterized by filament aggregation of tau protein with the displacement of the nucleus to the periphery part of the soma, and (3) the extracellular tangles, or the ghost NFTs stage, that results from a neuronal loss due to large amounts of filamentous tau protein with partial resistance to proteolysis [ 24 , 25 ].
3.3. Synaptic Loss
A synaptic damage in the neocortex and limbic system causes memory impairment and generally is observed at the early stages of AD. Synaptic loss mechanisms involve defects in axonal transport, mitochondrial damage, oxidative stress, and other processes that can contribute to small fractions, like the accumulation of Aβ and tau at the synaptic sites. These processes eventually lead to a loss of dendritic spines, pre-synaptic terminals, and axonal dystrophy [ 26 ]. Synaptic proteins serve as biomarkers for the detection of synapses loss, and severity, such as neurogranin, a postsynaptic neuronal protein, visinin-like protein-1 (VILIP-1), and synaptotagmin-1 [ 27 , 28 ].
4. The Stages of Alzheimer’s Disease
The clinical phases of Alzheimer’s disease can be classified into (1) pre-clinical or the pre-symptomatic stage, which can last for several years or more. This stage is characterized by mild memory loss and early pathological changes in cortex and hippocampus, with no functional impairment in the daily activities and absence of clinical signs and symptoms of AD [ 1 , 29 , 30 ]. (2) The mild or early stage of AD, where several symptoms start to appear in patients, such as a trouble in the daily life of the patient with a loss of concentration and memory, disorientation of place and time, a change in the mood, and a development of depression [ 30 , 31 ]. (3) Moderate AD stage, in which the disease spreads to cerebral cortex areas that results in an increased memory loss with trouble recognizing family and friends, a loss of impulse control, and difficulty in reading, writing, and speaking [ 30 ]. (4) Severe AD or late-stage, which involves the spread of the disease to the entire cortex area with a severe accumulation of neuritic plaques and neurofibrillary tangles, resulting in a progressive functional and cognitive impairment where the patients cannot recognize their family at all and may become bedridden with difficulties in swallowing and urination, and eventually leading to the patient’s death due to these complications [ 1 , 32 ].
5. Causes and Risk Factors of Alzheimer’s Disease
AD has been considered a multifactorial disease associated with several risk factors ( Figure 2 ) such as increasing age, genetic factors, head injuries, vascular diseases, infections, and environmental factors (heavy metals, trace metals, and others). The underlying cause of pathological changes in Alzheimer’s disease (Aβ, NFTs, and synaptic loss) is still unknown. Several hypotheses were proposed as a cause for AD but two of them are believed to be the main cause: some believe that an impairment in the cholinergic function is a critical risk factor for AD, while others suggest that alteration in amyloid β-protein production and processing is the main initiating factor. However, at present, there is no accepted theory for explaining the AD pathogenesis [ 33 , 34 ].

The risk factors for Alzheimer’s disease.
5.1. Alzheimer’s Disease Hypotheses
5.1.1. cholinergic hypothesis.
In the 1970s, neocortical and presynaptic cholinergic deficits were reported to be related to the enzyme choline acetyltransferase (ChAT), which is responsible for the synthesis of acetylcholine (ACh). Due to the essential role of ACh in cognitive function, a cholinergic hypothesis of AD was proposed. ACh is synthesized in the cytoplasm of cholinergic neurons from choline and acetyl-coenzyme A by the ChAT enzyme and transported to the synaptic vesicles by vesicular acetylcholine transporter (VAChT) ( Figure 3 ). In the brain, ACh is involved in several physiological processes such as memory, attention, sensory information, learning, and other critical functions. Degeneration of the cholinergic neurons was found to take place in AD and to cause alternation in cognitive function and memory loss. Β -amyloid is believed to affect cholinergic neurotransmission and to cause a reduction in the choline uptake and a release of ACh. Studies demonstrated that cholinergic synaptic loss and amyloid fibril formation are related to Aβ oligomers’ neurotoxicity and to interactions between AChE and Aβ peptide. Additional factors also contribute to the progression of AD, such as a reduction in nicotinic and muscarinic (M2) Ach receptors, located on presynaptic cholinergic terminals, and the deficit in excitatory amino acid (EAA) neurotransmission, where glutamate concentration and D-aspartate uptake are significantly reduced in many cortical areas in AD brains. This is in addition to the use of cholinergic receptor antagonists such as scopolamine, which was found to induce amnesia. This effect can be reversed by using compounds that activate acetylcholine formation [ 35 , 36 , 37 ].

The pathway for the synthesis and transportation of acetylcholine between presynaptic and postsynaptic nerve terminals.
As a result, the cholinergic hypothesis is based on three concepts: reduced presynaptic cholinergic markers in the cerebral cortex, severe neurodegeneration of nucleus basalis of Meynert (NBM) in the basal forebrain, which is the source of cortical cholinergic innervation, and the role of cholinergic antagonists in memory decline compared to the agonists, which have the opposite effect [ 38 ].
5.1.2. Amyloid Hypothesis
For decades, it was recognized that abnormal deposition of β-sheets in the central nervous system has a strong correlation with dementia, which led to the concept of the amyloid hypothesis. However, it was found that the amyloid plaques (AP) also deposit in normal healthy brains with aging, which raised the question of whether AP deposition is responsible for AD onset or not? Therefore, in the recent years, alternative hypotheses were proposed for the non-inherited form of AD (NIAD), but at present, the amyloid hypothesis remains the most accepted pathological mechanism for inherited AD (IAD). The amyloid hypothesis suggests that the degradation of Aβ, derived from APP by β- and γ-secretase, is decreased by age or pathological conditions, which leads to the accumulation of Aβ peptides (Aβ40 and Aβ42). Increasing the ratio of Aβ42/Aβ40 induces Aβ amyloid fibril formation, resulting in neurotoxicity and tau pathology induction, and consequently, leading to neuronal cell death and neurodegeneration. AD risk factors and mutations of several genes like APP, PSEN1, and PSEN2 were found to affect Aβ catabolism and anabolism, which rapidly cause an accumulation of Aβ and fast progression of neurodegeneration [ 39 , 40 , 41 ].
5.2. Alzheimer’s Disease Risk Factors
5.2.1. aging.
The most important risk factor in AD is aging. Younger individuals rarely have this disease, and most AD cases have a late onset that starts after 65 years of age [ 42 ]. Aging is a complex and irreversible process that occurs through multiple organs and cell systems with a reduction in the brain volume and weight, a loss of synapses, and ventricles’ enlargement in specific areas accompanied by SP deposition and NFT. Moreover, several conditions might emerge during aging such as glucose hypometabolism, cholesterol dyshomeostasis, mitochondria dysfunction, depression, and cognitive decline. These changes also appear in normal aging, which makes it difficult to distinguish the cases in early AD [ 43 , 44 ]. AD can be divided based on age of onset into early-onset AD (EOAD), the rare form with around 1–6% of cases, in which most of them are familial AD characterized by having more than one member in more than one generation with AD, and ranges from 30–60 or 65 years. The second type is the late-onset AD (LOAD), which is more common with age of onset above 65 years. Both types may occur in people who have a family with a positive history of AD and families with a late-onset disease [ 45 ].
5.2.2. Genetics
Genetic factors were discovered over the years and were found to play a major role in the development of AD. 70% of the AD cases were related to genetic factors: most cases of EOAD are inherited in an autosomal dominant pattern and mutations in the dominant genes such as Amyloid precursor protein (APP) , Presenilin-1 (PSEN-1), Presenilin-2 (PSEN-2) , and apolipoprotein E (ApoE) are associated with AD [ 46 , 47 ].
Herein, we discuss the strong genetic risk factors in AD.
- Amyloid Precursor Protein (APP)
APP is a type I transmembrane protein cleaved by α-, β-, and γ-secretase to release Aβ and other proteins and is encoded by the APP gene on chromosome 21. Thirty mutations have been found in the APP gene in which twenty-five of them are related to AD and cause an accumulation of Aβ with elevated amounts. Meanwhile, there is one protective mutation, A673T, which protects against AD by decreasing Aβ, Aβ40, and Aβ42 secretion [ 48 , 49 ]. All mutations surround the secretase cleavage site, for example, the KM670/671NL mutation in mouse models has shown an increasing level of amyloid plaques in the hippocampus and cortex with no NFTs. A673V, D678H, D678N, E682K, and K687N mutations have shown cortical atrophy, whereas E682K has shown hippocampal atrophy. Neuropathological reports for the A673V mutation demonstrated a presence of NFTs and Aβ, activation of microglia and astrocytes, and neuronal loss, compared to the rest of the mentioned mutations, which show no change in the intracellular Aβ according to neuropathological reports [ 48 , 50 ]. Other mutations such as T714I, V715A, V715M, V717I, V717L, L723P, K724N, and I716V affect the γ-secretase cleavage site and cause an increase in the Aβ42/Aβ40 ratio, while E693G, E693K, D694N, and A692G mutations affect the α-secretase cleavage site and cause polymorphic aggregates with the ability to disrupt bilayer integrity. Also, the E693delta is a deletion mutation that enhances the formation of synaptotoxic Aβ [ 51 , 52 ].
- Presenilin-1 (PSEN-1) and Presenilin-2 (PSEN-2)
PSEN1 and PSEN2 genes are also the autosomal dominant form of EOAD located on chromosomes 14 and 1, respectively. PSEN-2 and PSEN-1 are homologous, with 67% similarity, with a difference in the N -terminus and the hydrophilic region. Mutation in PSEN1 gene is more common, with more than 200 mutations, while a rare form with less than 40 mutations was identified in the PSEN2 gene [ 53 , 54 ].
PSEN1 is a core protein that activates the γ-secretase complex and plays an important role in the production of Aβ from APP. Knockout studies of PSEN1 showed synaptic dysfunction and memory impairment in mice, which indicate its essential role in maintaining memory and neurons [ 51 ]. PSEN1 mutations are simple ones which include single amino acid substitution, and severe mutation can result from the substitutions of two amino acids [ 55 ]. Mutations in the PSEN1 gene increase the ratio of Aβ42/Aβ40 by decreasing Aβ40 levels. The results obtained by Sun et al. study demonstrated that C410Y or L435F mutations in PSEN1 knock-in mice increased the Aβ42/Aβ40 ratio due to a greater reduction in Aβ40 [ 56 ].
In contrast, PSEN-2 mutations are rare and play a minor role in Aβ production. Any mutation in PSEN-2 might have a severe effect on the Aβ 42/40 ratio, causing familial AD in the presence of normal PSEN-1 alleles. Some of the PSEN-2 mutations cause a significant increase in γ-secretase activity with an elevation in the Aβ-42 and Aβ 42/40 ratio level, such as N141I, T122P, M239V, and M239I, while others are rare polymorphisms and have no effect on Aβ-42, -40, and Aβ 42/40 ratio levels and are not considered as pathogenic mutations [ 53 , 57 ].
- Apolipoprotein E (ApoE)
ApoE protein is a glycoprotein expressed highly in the liver and brain astrocytes and some microglia and serves as a receptor-mediated endocytosis ligand for lipoprotein particles like cholesterol, which is essential for myelin production and normal brain function. The ApoE gene located on chromosome 19 has three isoforms, ApoE2, ApoE3, and ApoE4, due to single-nucleotide polymorphisms (SNPs) which cause changes in the coding sequence. The ApoEε4 allele is a strong risk factor for both EOAD and LOAD compared to ApoEε2 and ApoEε3 alleles that are associated with a lower risk and protective effect, respectively [ 58 ]. ApoEε4 plays an important role in Aβ deposition as a senile plaque and causes cerebral amyloid angiopathy (CAA), which is known as a marker for AD [ 59 ]. ApoEε4 was also shown to be associated with vascular damage in the brain, which leads to AD pathogenesis [ 60 ].
- ATP Binding Cassette Transporter A1 (ABCA1)
Adenosine triphosphate (ATP)-binding cassette transporter A1 (ABCA1) is part of a large ABC transporters family that regulate cholesterol efflux in the circulation, like apolipoproteins-AI (ApoAI), and into the brain, like ApoE. In addition, ABCA1 maintains the stability of ApoE lipidation and serves as a mediator for high-density lipoprotein (HDL) generation, which reflects its role in atherosclerosis and cardiovascular diseases. Studies on the AD mice model showed that ABCA1 deficiency increases amyloid plaques and eliminates the lipidation of ApoE [ 61 ]. In humans, a mutation in ABCA1 results in Tangier disease, which is characterized by low levels of high-density lipoprotein (HDL) and ApoAI in plasma, accumulation of cholesterol in tissues, and AD pathogenesis [ 62 ].
- Clusterin Gene (CLU) and Bridging Integrator 1 ( BIN1 )
In contrast to PSEN1 , PSEN2 , and APP mutations, which result in familial or EOAD, clusterin ( CLU) and Bridging Integrator 1 ( BIN1 ) genes are novel risk factors for LOAD. In 2009, Genome-Wide Association Studies (GWAS) identified the CLU gene located on chromosome 8, which is upregulated in the cortex and hippocampus of AD brains, in addition to AD cerebrospinal fluid (CSF) and plasma, which make the CLU a promising biomarker for AD. The CLU may play a protective role by interacting with Aβ and promoting its clearance, or a neurotoxic role by reducing Aβ clearance. The Aβ ratio values determine whether the CLU role is neuroprotective or neurotoxic [ 63 ].
BIN1 is a Bin-Amphiphysin-Rvs (BAR) adaptor protein that is involved in the production of membrane curvature and other endocytosis cellular functions. BIN1 has several isoforms: some are found in the brain, where they interact with different proteins such as clathrin, synaptojanin, and amphiphysin 1, and others in which they regulate synaptic vesicle endocytosis. Recently, BIN1 was recognized as the second most important risk factor for LOAD after ApoE, where it plays a role in Aβ production and as a tau and NFT pathology modulator [ 64 , 65 ].
- Evolutionarily Conserved Signaling Intermediate in Toll pathway (ECSIT)
A significant accumulation of Aβ in AD brains increases protein oxidation, which reflects the critical role of mitochondria in Aβ cytotoxicity and AD pathogenesis. Evolutionarily conserved signaling intermediate in Toll pathway (ECSIT) gene is located on chromosome 19 and is associated with increasing the risk of AD. ECSIT encodes the adapting protein that functions as a cytoplasmic and signaling protein and is responsible for stabilizing the mitochondrial respiratory complex. Moreover, the adaptor protein is involved in the activation of nuclear factor (NF)-κB, interferon regulatory factors (IRFs), and activating protein-1. Also, it is involved in coupling immune toll-like receptor (TLR), homeostatic bone morphogenetic pathway (BMP), and transforming growth factor-beta (TGF-b) pathways [ 66 , 67 ].
ECSIT interacts with mitochondrial proteins such as Lon protease homolog (LONP1) and glutaryl-CoA dehydrogenase (GCDH), which are involved in intra-mitochondrial proteolysis and redox signaling respectively, followed by interactions with AD seed nitric oxide synthase (NOS3). Moreover, studies have shown certain interactions of ECSIT with the AD genes ApoE , PSEN-1 , and PSEN-2 . These interactions support the role of ECSIT as a molecular link in oxidative stress, inflammation, and mitochondrial dysfunction in AD [ 66 , 68 ].
- Estrogen Receptor Gene (ESR)
AD affects both women and men, but nearly two-thirds of AD cases are women. Several studies have shown that women with AD experience worse mental deterioration than men. Additionally, on the genetic level, some genes’ variation, like the ApoE4 allele, significantly increases AD risk in women compared to men. Other studies documented that AD risk in women is associated with the loss of ovarian hormones during menopause due to the fact that estrogen regulates several activities in the brain, such as neurotransmission, neural development, survival, protection against oxidative stress, reduction of Aβ peptide levels, and attenuation of tau hyperphosphorylation. The estrogen activity is mediated through estrogen receptors (ERs) (intracellular, transmembrane, and membrane-bound ERs). The two major subtypes of these receptors are ERα and Erβ, which are encoded by two distinct genes and are located on chromosome 6 and 14, respectively. ERα receptor is found in the hypothalamus and amygdala, whereas ERβ receptors are in the hippocampus and cortex. Single nucleotide polymorphisms (SNPs) in ERβ and ERα genes may affect exogenous estrogen in older women and influence cognitive aging. PvuII (rs9340799) and Xbal (rs223493) are examples of SNPs found in ERα and are associated with AD and cognitive impairment. Also, several SNPs in ERβ have been proven to increase the risk of AD in women [ 69 , 70 , 71 , 72 ].
- Other Genes
Other genes’ polymorphism associated with increasing the risk of AD include vitamin D receptor (VDR) gene polymorphism, which affects the affinity of vitamin D to its receptor and may cause neurodegenerative diseases and neuronal damage [ 73 ]. Moreover, epigenetic factors like DNA methylation, histone, and chromatin modifications were demonstrated to be involved in AD [ 33 , 74 ].
5.2.3. Environmental Factors
Aging and genetic risk factors cannot explain all cases of AD. Environmental risk factors including air pollution, diet, metals, infections, and many others may induce oxidative stress and inflammation and increase the risk for developing AD. Herein, we report the most important environmental factors and their relationships with AD [ 75 , 76 ].
- Air Pollution
The air pollution is characterized by modifying the nature of the atmosphere through the introduction of chemical, physical, or biological pollutants. It is associated with respiratory and cardiovascular diseases and recently, its association with AD was documented. Six air pollutants have been defined by National Ambient Air Quality Standards (NAAQSs) in the USA as a threat to human health, including ozone (O 3 ), nitrogen oxides (NO x ), carbon monoxide (CO), particulate matter (PM), sulfur dioxide (SO 2 ), and lead. Studies on animals and cellular models have shown that an exposure to high levels of air pollution can result in a damage to the olfactory mucosa and bulb, in addition to the frontal cortex region, similar to that observed in AD. In individuals exposed to air pollutants, there is a link between oxidative stress, neuroinflammation, and neurodegeneration, with the presence of hyper-phosphorylated tau and Aβ plaques in the frontal cortex. The air pollution can cause an increase in Aβ 42 formation, accumulation, and impaired cognitive function [ 77 , 78 ].
In recent years, the number of studies on the role of nutrition in AD have been increased. Several dietary supplements such as antioxidants, vitamins, polyphenols, and fish were reported to decrease the risk of AD, whereas saturated fatty acids and high-calorie intake were associated with increasing the risk of AD [ 79 ]. The food processing causes degradation of heat-sensitive micronutrients (e.g., vitamin C and folates), loss of large amounts of water, and formation of toxic secondary products (advanced glycation end products, AGEs) from non-enzymatic glycation of free amino groups in proteins, lipids, and nucleic acids. The toxic effect of AGEs is referred to as their ability to induce oxidative stress and inflammation by modifying the structure and function of the cell surface receptors and body proteins. Different studies demonstrated that elevated AGEs serum level is associated with cognitive decline and progression of AD. The AGE receptor (RAGE) is located in different places within the body, including microglia and astrocytes, and was established to be overexpressed in the brain of AD patients and serve as a transporter and a cell surface receptor for Aβ [ 80 ]. Malnutrition is another risk factor for AD. Deficiency in nutrients such as folate, vitamin B12, and vitamin D may cause a decrease in cognitive function, in addition to the fact that patients with AD suffer from problems associated with eating and swallowing, which may increase the risk of malnutrition [ 81 ].
Metals are found in nature and biological systems and can be divided into bio-metals that have a physiological function in living organisms (e.g., copper, zinc, and iron), and toxicological metals which do not possess any biological function (e.g., aluminum and lead) [ 82 ]. Aluminum is used significantly in the industries such as processed foods, cosmetics, medical preparations, medicines, and others. In the body, aluminum is bound to plasma transferrin and to citrate molecules that can mediate the transfer of aluminum to the brain. Studies demonstrated that Al accumulates in the cortex, hippocampus, and cerebellum areas, where it interacts with proteins and causes misfolding, aggregation, and phosphorylation of highly phosphorylated proteins like tau protein, characteristic of AD [ 83 ]. Lead competes with the binding site of bio-metals like calcium and can cross the blood–brain barrier (BBB) rapidly, where it can modify neural differentiation and synaptogenesis and cause severe damage. Studies revealed that an acute exposure to lead was associated with AD and caused an increase of β-secretase expression and Aβ accumulation. Cadmium is a carcinogenic water-soluble metal that can cross the BBB and cause neurological diseases like AD. Results have demonstrated that Cadmium ions are involved in the aggregation of Aβ plaques and the self-aggregation of tau in the AD brain. The data accumulated on metals support the notion that they are among the risk factors involved in the development of AD [ 84 ].
Chronic infections to the central nervous system (CNS) can cause an accumulation of Aβ plaques and NFT, therefore, they are included among the risk factors in AD. Studies by Dr. Itzhaki showed that the DNA of herpes simplex virus (HSV-1) was found in patients with ApoE-ε4 allele carriers, which explains the high risk for developing AD. HSV-1 can replicate in the brain, which can result in the activation of the inflammatory response and an increase in Aβ deposition, resulting in damage to neurons and gradual development of AD. On the other hand, the study results by Miklossy and Balin’s have revealed the role of chronic bacterial infections in AD. For example, syphilitic dementia caused by spirochete bacteria ( Treponema pallidum ), which are accumulated in the cerebral cortex, produced lesions similar to neurofibrillary tangles, which led to devastating neurodegenerative disorders. Besides, Chlamydia pneumonia bacterium can trigger late-onset AD by activation of astrocyte and cytotoxic microglia, disrupt calcium regulation and apoptosis, resulting in deterioration of cognitive function, and increase the risk of AD [ 85 , 86 , 87 ].
5.2.4. Medical Factors
Several risk factors are related to the development of Alzheimer’s disease. Adding to this list, older people with AD usually have medical conditions such as cardiovascular disease (CVD), obesity, diabetes, and others. All of these conditions are associated with increased risk of AD [ 88 , 89 ].
- Cardiovascular Disease (CVDs)
CVDs are recognized as an important risk factor for AD, such as the stroke that is associated with increased risk of dementia due to a neural tissue loss, which enhances degenerative effect and influences amyloid and tau pathology. Atrial fibrillation also causes embolisms which leads to stroke and a decrease in memory and cognitive functions. Moreover, heart failure affects the pumping function of the heart and results in insufficient blood supply to the body and hypo-perfusion of the brain that leads to hypoxia and neural damage. The coronary heart disease’s hypothesis indicates that atherosclerosis, peripheral artery disease, hypo-perfusion, and emboli are all related to increased risk of AD. Hypertension is associated with thickening of vessel walls and narrowing of the lumen which reduce the cerebral blood flow, and in chronic cases, it may cause cerebral edema, which all participate as risk factors for AD and CVD. The CVD is a modifiable risk factor and by focusing on its relationship with AD, a pathway to prevent and delay the disease can be obtained [ 89 , 90 ].
- Obesity and Diabetes
Obesity is a term used for too much body fat in individuals due to consuming more calories than they burn and can be calculated by using the body mass index (BMI). Increasing the body fat is associated with a decreased brain blood supply which promotes brain ischemia, memory loss, and vascular dementia. The obesity, unhealthy diet, and other factors can cause impaired glucose tolerance (IGT) or diabetes, which is characterized by hyperglycemia that affects peripheral tissues and blood vessels. Chronic hyperglycemia can induce cognitive impairment as a result of increasing amyloid-beta accumulation, oxidative stress, mitochondrial dysfunction, and neuroinflammation. Obesity is characterized by increasing pro-inflammatory cytokines secretions from adipose tissue, which stimulate macrophages and lymphocytes and eventually lead to local and systemic inflammation. This inflammation promotes insulin resistance, hyperinsulinemia, and as a consequence, hyperglycemia. Obesity is a well-known risk factor for type 2 diabetes, CVDs, and cancer, which are identified as risk factors for dementia and AD. The brain inflammation causes an increase in microglia and results in reduced synaptic plasticity and impaired neurogenesis. Microglia can affect insulin receptor substrate 1 (IRS-1) and block intracellular insulin signaling, which has an important role in neural health. Therefore, alteration in insulin action can result in Aβ accumulation and reduce the tau protein degradation associated with AD [ 91 , 92 , 93 , 94 ].
6. Treatment
Currently, Alzheimer’s disease cases worldwide are reported to be around 24 million, and in 2050, the total number of people with dementia is estimated to increase 4 times. Even though AD is a public health issue, as of now, there is only two classes of drugs approved to treat AD, including inhibitors to cholinesterase enzyme (naturally derived, synthetic and hybrid analogues) and antagonists to N -methyl d -aspartate (NMDA). Several physiological processes in AD destroy Ach-producing cells which reduce cholinergic transmission through the brain. Acetylcholinesterase inhibitors (AChEIs), which are classified as reversible, irreversible, and pseudo-reversible, act by blocking cholinesterase enzymes (AChE and butyrylcholinesterase (BChE)) from breaking down ACh, which results in increasing ACh levels in the synaptic cleft [ 95 , 96 , 97 ]. On the other hand, overactivation of NMDAR leads to increasing levels of influxed Ca 2+ , which promotes cell death and synaptic dysfunction. NMDAR antagonist prevents overactivation of NMDAR glutamate receptor and hence, Ca 2+ influx, and restores its normal activity. Despite the therapeutic effect of these two classes, they are effective only in treating the symptoms of AD, but do not cure or prevent the disease [ 98 , 99 ]. Unfortunately, only a few clinical trials on AD have been launched in the last decade and their outcome was a big failure. Several mechanisms have been proposed to understand AD pathology in order to modify its pathway and develop successful treatments, which include abnormal tau protein metabolism, β-amyloid, inflammatory response, and cholinergic and free radical damage [ 30 , 100 ]. On the other hand, most AD modifiable risk factors such as cardiovascular or lifestyle habits can be prevented without medical intervention. Studies showed that physical activity can improve the brain health and reduce AD by activating the brain vascularization, plasticity, neurogenesis, and reducing inflammation by decreasing Aβ production, which all result in improving cognitive function in older people. Moreover, the Mediterranean diet (MD), intellectual activity, and higher education all may reduce the progression of AD and memory loss and increase the brain capacity and cognitive functions. Several studies revealed that multi-domain intervention which includes lifestyle (diet, exercise, and cognitive training), depression of AD symptoms, and controlling cardiovascular risk factors, can increase or maintain cognitive function and prevent new cases of AD in older people [ 101 ]. Herein, we summarize the currently available drugs and theories for the development of new therapies for AD.
6.1. Symptomatic Treatment of AD
6.1.1. cholinesterase inhibitors.
According to the cholinergic hypothesis, AD is due to the reduction in acetylcholine (ACh) biosynthesis. Increasing cholinergic levels by inhibiting acetylcholinesterase (AChE) is considered one of the therapeutic strategies that increases cognitive and neural cell function. AChEIs are used to inhibit acetylcholine degradation in the synapses, which results in continuous accumulation of ACh and activation of cholinergic receptors. Tacrine (tetrahydroaminoacridine) ( 1, Figure 4 ) was the first FDA (Food and Drug Administration)-approved cholinesterase inhibitor drug for the treatment of AD, which acts by increasing ACh in muscarinic neurons, but it exited the market immediately after its introduction due to a high incidence of side effects like hepatotoxicity and a lack of benefits, which was observed in several trials. Later on, several AChEIs were introduced, such as donepezil ( 2 , Figure 4 ), rivastigmine ( 3 , Figure 4 ), and galantamine ( 4 , Figure 4 ), and are currently in use for the symptomatic treatment of AD [ 34 , 97 , 102 , 103 ]. Another strategy that may help in the treatment of AD is increasing choline reuptake and as a result, increasing acetylcholine synthesis at the presynaptic terminals. This can be achieved by targeting choline transporter (CHT1) which is responsible for supplying choline for the synthesis of ACh. Developing drugs that are capable of increasing CHT1 at the plasma membrane may become the future therapy of AD [ 36 ].

The chemical structures of approved drugs for symptomatic treatment of AD (tacrine 1 , donepezil 2 , rivastigmine 3 , galantamine 4 , and memantine 5 ) and disease-modifying compounds that entered clinical trials (semagacestat 6 , avagacestat 7 , tarenflurbil 8 , lanabecestat 9 , verubecestat 10 , atabecestat 11 , umibecestat 12 , methylene blue 13 , tideglusib 14 , and saracatinibin 15 ).
Donepezil ( 2 , Figure 4 ) is an indanonebenzylpiperidine derivative and a second generation of AChEIs and is considered the leading drug for AD treatment. Donepezil binds to acetylcholinesterase reversibly and inhibits acetylcholine hydrolysis, which leads to a higher concentration of ACh at the synapses. The drug is well-tolerated with mild and transient cholinergic side effects which are related to the gastrointestinal and nervous systems. It should be noted that donepezil is used to treat symptoms of AD such as improving cognition and behavior without altering the AD progression [ 104 , 105 , 106 ].
- Rivastigmine
Rivastigmine ( 3 , Figure 4 ) is a pseudo irreversible inhibitor of AChE and butyrylcholinesterase (BuChE) that acts by binding to the two active sites of AChE (anionic and estearic sites), which results in preventing ACh metabolism. BuChE is found mostly in glial cells with only 10% of AChE activity in the normal brain, whereas in the AD brain, its activity is increased to 40–90%, while ACh activity is reduced simultaneously, which suggests that BuChE action may indicate a moderate to severe dementia. Rivastigmine dissociates more slowly than AChE, which is why it is called a pseudo-irreversible, and it undergoes metabolism at the synapse by AChE and BuChE. The drug is used in mild to moderate AD cases. It improves cognitive functions and daily life activities. Oral administration of the drug is associated with adverse effects such as nausea, vomiting, dyspepsia, asthenia, anorexia, and weight loss. In many cases, these side effects are the main reason behind stopping taking the medicine, however, they can be settled down in time and consequently, the drug becomes more tolerated. Rivastigmine can be delivered by transdermal patches for controlled and continuous delivery of the drug through the skin, with enhanced tolerability and caregiver satisfaction. Also, the patches can deliver a lower dosage compared to pills, which results in reduced side effects. Most AD patients suffer from memory loss and swallowing problems which affect their compliance in administering oral drugs at regular intervals. Therefore, the use of transdermal patches is the most appropriate method for delivering the drug in AD patients [ 107 , 108 , 109 , 110 ].
- Galantamine (GAL)
Galantamine ( 4 , Figure 4 ) is considered a standard first-line drug for mild to moderate AD cases. GAL is a selective tertiary isoquinoline alkaloid with a dual mechanism of action in which it acts as a competitive inhibitor of AChE and can bind allosterically to the α-subunit of nicotinic acetylcholine receptors and activate them. GAL can improve behavioral symptoms, daily life activities, and cognitive performance with good efficacy and tolerability, similar to other AChE inhibitors. Several delivery systems were developed to improve the drug delivery to the brain: Wahba et al. attached GAL to ceria-containing hydroxyapatite particles for selective delivery of the drug to the affected regions in the brain. Misra et al. and Fornaguera et al. used solid-lipid nanoparticles and nano-emulsification approaches respectively, to carry GAL hydrobromide. The results of these studies demonstrated a promising strategy for safe delivery of the drug. Hanafy et al. developed nasal GAL hydrobromide/chitosan complex nanoparticles which showed good pharmacological efficacy, while Woo et al. utilized the patch system as a carrier for a controlled release dosage form of the drug [ 111 , 112 , 113 , 114 ].
6.1.2. N -methyl d -aspartate (NMDA) Antagonists
NMDAR is believed to have a dominant role in the pathophysiology of AD. NMDAR stimulation results in Ca 2+ influx which activates signal transduction and as a consequence, it triggers gene transcription essential for the formation of a long-term potentiation (LTP), which is important for synaptic neurotransmission, plasticity, and memory formation. Over-activation of NMDARs causes an abnormal level of Ca 2+ signaling and overstimulation of glutamate, which is the primary excitatory amino acid in the CNS, which results in excitotoxicity, synaptic dysfunction, neuronal cell death, and a decline in cognitive functions. Several NMDAR uncompetitive antagonists have been developed and entered clinical trials, however, most of them failed due to low efficacy and side effects. Memantine ( 5 , Figure 4 ) is the only approved drug in this category to treat moderate to severe AD; in addition, other NMDAR uncompetitive antagonist compounds are being developed, such as RL-208 (3,4,8,9-tetramethyltetracyclo [4.4.0.0 3,9 .0 4,8 ]dec-1-yl)methylamine hydrochloride), a polycyclic amine compound that may possess a promising therapeutic effect in age-related cognitive problems and AD [ 115 , 116 , 117 ].
Memantine ( 5 , Figure 4 ) is a low-affinity uncompetitive antagonist of the NMDAR, a subtype of glutamate receptor that prevents over-activation of the glutaminergic system involved in the neurotoxicity in AD cases. Memantine is used for the treatment of moderate to severe AD alone or in combination with AChEI. The drug is safe and well-tolerated, it blocks the excitatory receptor without interfering with the normal synaptic transmission due to memantine’s low affinity, where it is displaced rapidly from NMDAR by high concentrations of glutamate, thus avoiding a prolonged blockage. The latter is associated with high side effects, especially on learning and memory [ 99 , 118 ].
6.2. Promising Future Therapies
6.2.1. disease-modifying therapeutics (dmt).
Disease-modifying treatment or therapy (DMT) alter the progression of AD by working on several pathophysiological mechanisms. This is in contrast to symptomatic therapy which works on improving the cognitive functions and decreasing symptoms such as depression or delusions without affecting or modifying the disease. DMTs, either immunotherapies or small molecules, are administrated orally and are being developed to prevent AD or decrease its progression. Several DMTs have been developed and entered the clinical trials, such as AN-1792, a synthetic Aβ peptide (human Aβ 1–42 peptide of 42-amino acids with the immune adjuvant QS-21) and the first active immunotherapy for AD which entered phase II clinical trials and discontinued due to a meningoencephalitis side effect in 6% of the patients. Other drugs were also developed and failed in the clinical trials, including the anti-Aβ antibody (solanezumab and bapineuzumab), γ-Secretase inhibitors (semagacestat 6 , avagacestat 7 , and tarenflurbil 8 ) ( Figure 4 ) and β-secretase inhibitors (BACE) (Lanabecestat 9, verubecestat 10 , and atabecestat 11 ) ( Figure 4 ). DMTs failures are due to several factors, such as starting therapy too late, giving treatment for the wrong main target, use of inappropriate drug doses, and misunderstanding of the pathophysiology of AD. Several immunotherapies described in Table 1 have been developed over decades, including: CAD106, an active Aβ immunotherapy that induces Aβ antibodies in animal models and consists of multiple copies of Aβ1–6 peptide coupled to Qβ coat protein, a virus-like particle, and is still in clinical trials, and CNP520 (umibecestat, 12 ) ( Figure 4 ), a small molecule that inhibits beta-scretase-1 (BACE-1) and therefore inhibits Aβ production. CNP520 was found to reduce Aβ plaque deposition and Aβ levels in the brain and CSF in rats, dogs, and healthy adults ≥ 60 years old, and is still under clinical trials. Furthermore, aducanumab, gantenerumab, and crenezumab are all human Aβ monoclonal antibody that bind with high affinity to aggregated Aβ, and they are still under study in the clinical phases with other DMTs described in Table 1 [ 6 , 119 , 120 , 121 , 122 , 123 , 124 ].
Disease modifying agents for the treatment of Alzheimer’s disease in clinical trials.
Another class targeting the α-secretase enzyme was developed and has been considered as therapeutic agents. α-secretase modulators or activators stimulate the cleavage of APP. There is little knowledge about the activation pathway, but research assumes that it is promoted by the phosphatidylinositol 3-kinase (PI3K)/Akt pathway or by γ-aminobutyric acid (GABA) receptor signaling. Targeting these pathways may give potential therapeutic agents for AD [ 6 ].
In addition to the anti-amyloid agents, the tau aggregation inhibitors are another promising DMT. The tau is a biomarker for neurofibrillary tangles (NFT) in AD and naturally modulates microtubule stability, signaling pathways, and axonal transport. A modification in tau conformation results in toxic aggregation. Therefore, the prevention of tau aggregation becomes an interesting approach for drug discovery to reduce AD progression. Studies in mice have shown that tau oligomers cause mitochondrial damage, disruption of neuronal signaling, synaptic loss, and memory impairment. Disease-modifying therapeutics (DMT) like small molecules can be used to inhibit the initial step in the tau aggregation and thereby reduce its accumulation. Methylene blue ( 13 , Figure 4 ) is a blue dye that inhibits the tau aggregation and entered phase II clinical trials to treat mild to moderate AD. Upon administration of the drug, the color of the urine becomes blue, which indicates a lack of binding, and because of that, the study was highly criticized. Other approaches suggest that an inhibition of specific kinases such as glycogen synthase kinase 3 (GSK3β) can inhibit tau hyperphosphorylation and block tau deposition. Examples of these entities include tideglusib ( 14 , or NP-031112 (NP-12), Figure 4 ), a thiazolidinedione-derived compound, lithium, pyrazolopyridines, pyrazolopyrazines, sodium valproate, and others. Another protein kinase inhibitor is saracatinib (AZD0530) ( 15 , Figure 4 ), which acts by inhibiting tyrosine kinase and has shown good results in improving memory in transgenic mice and is currently in phase II trials [ 125 , 126 , 127 ]. Davidowitz et al. utilized the hatu mouse model of tauopathy to study the efficacy of a lead small molecule in preventing tau accumulation. The study results demonstrated a significant reduction in tau levels and its phosphorylated form levels, which indicates the ability to inhibit the entire pathway of the tau aggregation by using an optimized lead compound [ 128 ].
6.2.2. Chaperones
Protein misfolding caused by mutations or environmental factors results in aggregations that are toxic, and their accumulation causes neurodegenerative disorders like AD. Naturally, cells develop protein quality control (PQC) systems that inhibit protein misfolding before exerting their toxic effects. With age, this balance is altered and the misfolded shapes overwhelm the PQC system, which in turn activates the unfolded protein response (UPR) that stops the protein synthesis and increases chaperone production. Generally, the cells in humans have proteins that are responsible for other proteins to function and arrive to their destination in the cell. These proteins are called “chaperones”. Chaperones are involved in protein folding and improvement of the PQC system efficiency. Therefore, it is considered a promising candidate for treating neurodegenerative diseases. It can be classified into three groups: (1) molecular chaperones, which are proteins that assist other nonnative proteins in their folding or unfolding, like overexpression of heat shock proteins (Hsps) that serve as neuroprotective agents, (2) pharmacological chaperones, which are low molecular weight compounds (enzymes or receptor-ligand or selective binding molecules) that induce refolding of proteins, stabilize their structure, and restore their function, and (3) chemical chaperones, also low molecular weight compounds, which are divided into two groups, osmolytes and hydrophobic compounds. The members in these two groups have no specific mechanism of action and need high concentrations to exert their therapeutic effects [ 129 ].
- Heat Shock Proteins (Hsps)
The causes for most neurodegenerative diseases are protein misfolding and aggregation, which lead to cell death. The molecular chaperone can be intracellular, such as in the case of heat shock proteins (e.g., Hsp40, Hsp60, Hsp70, Hsp90, Hsp100, and Hsp110), and extracellular, such as clustering and alpha-macroglobulin. HSPs play an essential role in the protein folding process and protect cells from harmful stress-related events. There are two families of Hsps: (a) classic Hsps that possess an ATP-binding site with a molecular weight of 60 kD or more. This family includes Hsp100, Hsp90, Hsp70, and Hsp60, and (b) the small Hsps such as αB-crystalline, Hsp27, Hsp20, HspB8, and HspB2/B3 that lack ATP-binding site, with a molecular weight of 40 kD or less. These proteins can assist other Hsps in their refolding function. Failure of these mechanisms can lead to oxidative stress, mitochondrial dysfunction, and many other conditions that cause damage, a loss of neurons, and a progression of neurodegenerative diseases. Different HSPs can block the aggregation process of misfolded proteins, like amyloidogenic proteins (Aβ and tau), and promote their degradation [ 130 , 131 ].
Hsp60 plays an important role in mitochondrial protein folding. Its role in AD is not clear, some believe that the protein has a protective role and others think it has a harmful effect where it can be over-expressed by activated microglia, which increases pro-inflammatory factors such as toll-like receptor 4 (TLR-4) that stimulate neuronal cell death. Therefore, inhibiting activated microglia and Hsp60 expression is a promising strategy for preventing neurodegenerative diseases. Examples of compounds that inhibit Hsp60 are mizoribine (Immunosuppressant) ( 16 , Figure 5 ) and pyrazolopyrimidine EC3016 ( 17 , Figure 5 ). Both compounds act by blocking ATPase activity of Hsp60 and inhibiting protein folding. On the other hand, avrainvillamide, a fungal metabolite ( 18 , Figure 5 ), and epolactaene, a bacterial metabolite ( 19 , Figure 5 ), act by binding to the Hsp60′s cysteine residues and inhibit its folding activity. However, Hsp60’s role in AD remains controversial and there is a need for more investigations to understand its role [ 130 ].

The chemical structures of different chaperone molecules: Mizoribine 16 , EC3016 17 , Avrainvillamide 18 , Epolaztaene 19 , MKT-077 20 , YM-01 21 , JG-98 22 , Radicicol 23 , Geldanamycin 24 , 17-AAG 25 , Pochoxime C (OS47720) 26 , R55 27 , and OT1001 28 .
Studies have shown that Hsp70 binds to Aβ42 and prevents self-aggregation. Martín-Peña et al. studied two isoforms of Hsp70, cytosolic and extracellular, in Drosophila flies AD models and evaluated their protective role against memory decline that results from Aβ42 aggregation. The animal studies showed that Hsp70 has a dual function: intracellularly and extracellularly, where it protects against Aβ42 neurotoxicity and synaptic loss. In addition to its ability to bind to tau and its hyper-phosphorylated form and prevent its formation, it decreases aggregation and promotes tau binding to microtubules. Hsp70 acts by activating microglia, insulin-degrading enzyme, and tumor growth facto r- β1, which degrades β-amyloids and prevents memory impairments [ 132 , 133 ]. Some studies in AD brain tissue demonstrated an overexpression of Hsp70 levels and a correlation with the presence of activated glia and stressed neurons. Also, it was found that Hsp70 is associated with extracellular deposits in AD. Drug therapies targeting Hsp70, mainly referring to previous anticancer drugs which target and inhibit Hsp70 ATP-binding site, are considered as candidates in AD treatment due to their ability to reduce tau levels in vitro and ex vivo. MKT-077(1-ethyl-2-(( Z )-(( E )-3-ethyl-5-(3-methylbenzo [ d ]thiazol-2(3 H )-ylidene)-4-oxothiazolidin-2-ylidene)methyl)pyridin-1-ium chloride) ( 20 , Figure 5 ), is an anticancer rhodacyanine compound that binds to mortalin, a mitochondrial Hsp70 site, and acts as an anti-proliferative agent, but the use of this compound was stopped due to toxicity side effects and low BBB penetration. On the other hand, YM-01 ( 21 , Figure 5 ), a more potent MKT-077 derivative, was developed with a single replacement of the ethyl group on the pyridinium nitrogen of MKT-077 with a methyl group. JG-98 ( 22 , Figure 5 ) is also an MKT-077 derivative with a 60-fold higher binding affinity to Hsp70 than YM-01 [ 130 , 134 , 135 , 136 ].
Hsp90 is another type of HSP that regulates the tau phosphorylation and dephosphorylation. An inhibition of Hsp90 results in a decrease in phosphorylation of tau due to a reduction in tau kinases, which is thought to be responsible for tau pathogenesis when it is hyperactivated. Hsp90 inhibitors are used for cancer therapy, but recently, they are considered as promising therapy for AD. Radicicol (RDC) ( 23 , Figure 5 ) and geldanamycin (GA) ( 24 , Figure 5 ) are Hsp90 inhibitors. GA is a natural antifungal compound and the first discovered Hsp90 inhibitor. Studies on this inhibitor were stopped due to its toxicity. On the other hand, 17-AAG (17-(Allylamino)-17-demethoxygeldanamycin) ( 25 , Figure 5 ) is a GA derivative with a lower toxicity and better pharmacokinetic profile that showed a good improvement of the cognitive function by inducing other HSPs, like Hsp70, in addition to reducing NFTs in the transgenic mouse model by blocking the tau phosphorylation pathway, indirectly [ 137 , 138 ]. Pochoxime C (OS47720) ( 26 , Figure 5 ) is also a CNS-permeable Hsp90 inhibitor that showed good safety and efficacy profiles when tested in the AD mouse model. Studies revealed that OS47720 acts by strengthening synaptic function via heat shock factor (HSF-1) activation and dependent transcriptional events [ 139 ].
The combined studies demonstrate that targeting HSPs is a promising strategy to develop drugs with a new mechanism of action for reducing pathogenic tau levels and restoring normal tau homeostasis.
- Vacuolar sorting protein 35 (VPS35)
An accumulation of proteins in neurons and glial cells leads to disturbance of cellular protein homeostasis. The endosomal-lysosomal system is responsible for transporting proteins for recycling and degradation. Any malfunction in the system can lead to several diseases, such as Alzheimer’s disease. Retromer is a complex of regulator proteins composed of sorting nexin (SNX1, 2, 5, 6) and vacuolar sorting proteins (VPS 26, 29, 35), which are responsible for transporting cargo molecules from the endosome to the trans -Golgi network. A loss of retromer’s function results in the downregulation of VPS35, which can increase Aβ formation, induce cognitive impairments, and cause synaptic dysfunction, which is reported in AD patients [ 140 , 141 ]. A study on 3xTg mice brains was conducted to evaluate the effect of VPS35 overexpression on memory function. The study showed that a significant reduction of the Aβ peptide and tau neuropathology (soluble, insoluble, and phosphorylated tau) was associated with overexpression of VPS35, in addition to a reduction in neuroinflammation and ameliorating synaptic dysfunction [ 142 ]. Therefore, VPS35 is an important promising therapeutic target for AD treatment. A small pharmacological chaperones molecule called R55 (thiophene-2,5-diylbis(methylene) dicarbamimidothioatedihydrochloride) ( 27 , Figure 5 ), a thiophenethiourea derivative, can enhance retromer stability and function by increasing retromer proteins, shifting AOO from the endosome, and reducing pathogenic processing of APP, which may serve as a promising therapeutic molecule for neurodegenerative diseases [ 143 ].
Studies demonstrated that the accumulation of gangliosides has been associated with misfolding and aggregation of proteins in neurodegenerative diseases. Abnormal levels of mono-sialoganglioside (GM1, GM2, and GM3) have been reported in AD brains. Mutant forms of Aβ, like Dutch mutant APPE693Q, showed susceptibility to pro-aggregation properties of GM2 and GM3, resulting in the formation of Aβ peptides complexes with gangliosides (ganglioside-bound Aβ (GAβ) peptide) and subsequently leading to an acceleration of aggregation and accumulation of Aβ peptides.
β-hexosaminidase (β-hex) is a lysosomal enzyme that acts by catabolizing GM2 ganglioside, and increasing its activity can lead to a reduction of GM2 levels and Aβ aggregation and accumulation. Small molecules like pharmacological chaperones (PC) can selectively bind and stabilize wild-type proteins and restore their normal folding. OT1001 ( 28 , Figure 5 ) is an iminosugar PC that targets β-hex and increases its level in the brain and reduces GAβ pathology. Studies on Dutch APPE693Q transgenic mice showed that OT1001 has good pharmacokinetics, brain penetration ability, and tolerability, with lower side effects. These make the compound a good drug candidate for increasing the β-hex activity [ 144 ].
6.2.3. Natural Extract
For a long time, natural compounds have been used as therapeutic agents for several pathological diseases, and recent studies showed that they possess a neuroprotective effect. In vitro and in vivo studies have proven that natural compounds possess a therapeutic potential for AD, which allowed some of them to enter the clinical trials stages. Nicotine was the first natural compound entered in the clinical trials for AD, then other compounds like vitamins C, E, and D gained more attention and interest due to their protective role against neuroinflammation and oxidative damage. Recently, bryostatin, a macrolide lactone extract from bryozoan Bugula neritina, has been evaluated and showed the ability to induce α-secretase activity, reduce Aβ production, and enhance the learning and memory in an AD mice model [ 145 ]. Other natural compounds used in folk medicine (traditional Chinese medicine (TCM)) demonstrated a great potential in treating AD by acting on several mechanisms, as shown in Table 2 below [ 146 ].
Natural compounds used in folk medicine and their mechanism of actions.
7. Conclusions
Alzheimer’s disease is now considered a world health concern; as a consequence, the National Institute on Aging—Alzheimer’s Association reclassified and updated the 1984 NINCDS-ADRDA criteria for higher specificity, sensitivity, and early identification of patients at risk of developing AD. Several criteria have been proposed for a more accurate diagnosis of AD, including clinical biomarkers, bodily fluids, and imaging studies. Despite that, the treatment of AD remains symptomatic, without alteration in the disease’s prognosis. Inhibitors to cholinesterase enzyme such as galantamine, donepezil, and rivastigmine, and NMDA antagonists such as memantine, improve memory and alertness but do not prevent progression. Several studies have shown that modification in lifestyle habits like diet and exercise can improve brain health and reduce AD without medical intervention and is considered as a first-line intervention for all AD patients. Recently, the research is focusing on targeting the pathological features of AD such as Aβ and p-tau. Future therapies such as disease-modifying treatment can alter the progression of AD by targeting the Aβ pathway, and many drugs have entered the clinical trials, like AN-1792, solanezumab, bapineuzumab, semagacestat, avagacestat, and tarenflurbil, but failed in demonstrating efficacy in the final clinical stages. Other DMTs are still under investigation, such as those targeting Aβ and tau pathologies, such as aducanumab, gantenerumab, crenezumab, tideglusib, lithium, and others. Other promising compounds called chaperones like heat shock proteins and vacuolar sorting protein 35 (VPS35) function by assisting other proteins to function normally and to arrive at their destination in the cell safely, and therefore can be used as a treatment for neurodegenerative diseases. Moreover, the natural extracts used in folk Chinese medicine showed great potential in treating AD by acting on several mechanisms’ pathways. In conclusion, the success of AD treatment depends on its early administration and patient monitoring for disease progression using biomarkers diagnosis. Future therapies that target tau pathology and the use of combination therapy may have a potential to slow the progression of AD pathology. Designing a potent, selective, and effective drug is urgently needed to treat patients with AD and those at risk for developing the disease.
Author Contributions
Literature survey and first draft writing were done by Z.B., and final draft, including the revisions, were accomplished by R.K. All authors have read and agreed to the published version of the manuscript.
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Learn how UpToDate can help you.
Select the option that best describes you
- Medical Professional
- Resident, Fellow, or Student
- Hospital or Institution
- Group Practice
- Patient or Caregiver
- Find in topic
RELATED TOPICS
INTRODUCTION
This topic reviews the clinical manifestations and diagnosis of AD. Other topics review the risk factors and treatment of AD:
● (See "Epidemiology, pathology, and pathogenesis of Alzheimer disease" .)
● (See "Treatment of Alzheimer disease" .)
Other topics review the approach to patients with cognitive impairment and dementia and the clinical features of other dementia syndromes:
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Published: 13 May 2021
Alzheimer disease
- David S. Knopman ORCID: orcid.org/0000-0002-6544-066X 1 ,
- Helene Amieva 2 ,
- Ronald C. Petersen 1 ,
- Gäel Chételat 3 ,
- David M. Holtzman 4 ,
- Bradley T. Hyman ORCID: orcid.org/0000-0002-7959-9401 5 ,
- Ralph A. Nixon 6 , 7 &
- David T. Jones ORCID: orcid.org/0000-0002-4807-9833 1
Nature Reviews Disease Primers volume 7 , Article number: 33 ( 2021 ) Cite this article
42k Accesses
748 Citations
217 Altmetric
Metrics details
- Alzheimer's disease
- Diagnostic markers
- Translational research
Alzheimer disease (AD) is biologically defined by the presence of β-amyloid-containing plaques and tau-containing neurofibrillary tangles. AD is a genetic and sporadic neurodegenerative disease that causes an amnestic cognitive impairment in its prototypical presentation and non-amnestic cognitive impairment in its less common variants. AD is a common cause of cognitive impairment acquired in midlife and late-life but its clinical impact is modified by other neurodegenerative and cerebrovascular conditions. This Primer conceives of AD biology as the brain disorder that results from a complex interplay of loss of synaptic homeostasis and dysfunction in the highly interrelated endosomal/lysosomal clearance pathways in which the precursors, aggregated species and post-translationally modified products of Aβ and tau play important roles. Therapeutic endeavours are still struggling to find targets within this framework that substantially change the clinical course in persons with AD.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 1 digital issues and online access to articles
92,52 € per year
only 92,52 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Is Alzheimer disease a disease?

The Amyloid-β Pathway in Alzheimer’s Disease

Emerging diagnostics and therapeutics for Alzheimer disease
Jessen, F. et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement. 10 , 844–852 (2014).
PubMed PubMed Central Google Scholar
Petersen, R. C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 256 , 183–194 (2004).
CAS PubMed Google Scholar
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging and the Alzheimer’s Association workgroup. Alzheimers Dement. 7 , 263–269 (2011).
Petersen, R. C. How early can we diagnose Alzheimer disease (and is it sufficient)? The 2017 Wartenberg lecture. Neurology 91 , 395–402 (2018).
Nelson, P. T. et al. Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 121 , 571–587 (2011).
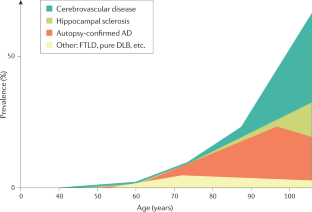
Boyle, P. A. et al. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann. Neurol. 83 , 74–83 (2018). A clinical-neuropathological analysis of >1,000 persons demonstrating how multiple aetiologies relate to late-life cognition.
CAS PubMed PubMed Central Google Scholar
Schneider, J. A., Arvanitakis, Z., Leurgans, S. E. & Bennett, D. A. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 66 , 200–208 (2009).
Kapasi, A., DeCarli, C. & Schneider, J. A. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 134 , 171–186 (2017).
Karanth, S. et al. Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. 77 , 1299–1307 (2020).
Brodaty, H. et al. The world of dementia beyond 2020. J. Am. Geriatr. Soc. 59 , 923–927 (2011).
PubMed Google Scholar
Wu, Y. T. et al. The changing prevalence and incidence of dementia over time — current evidence. Nat. Rev. Neurol. 13 , 327–339 (2017).
Satizabal, C. L. et al. Incidence of dementia over three decades in the Framingham Heart Study. N. Engl. J. Med. 374 , 523–532 (2016).
Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 11 , 1006–1012 (2012).
Tom, S. E. et al. Trends in incident dementia and early life socieoeconomic status by birth cohort in the adult changes in thought study. JAMA Open 3 , e2011094 (2020).
Google Scholar
Kovari, E., Herrmann, F. R., Bouras, C. & Gold, G. Amyloid deposition is decreasing in aging brains: an autopsy study of 1,599 older people. Neurology 82 , 326–331 (2014).
Niu, H., Álvarez-Álvarez, I., Guillén-Grima, F. & Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: a meta-analysis. Neurologia 32 , 523–532 (2017).
Hy, L. X. & Keller, D. M. Prevalence of AD among whites: a summary by levels of severity. Neurology 55 , 198–204 (2000).
Gillis, C., Mirzaei, F., Potashman, M., Ikram, M. A. & Maserejian, N. The incidence of mild cognitive impairment: a systematic review and data synthesis. Alzheimers Dement. 11 , 248–256 (2019).
Petersen, R. C. et al. Mild cognitive impairment due to Alzheimer’s disease: criteria in the community. Ann. Neurol. 74 , 199–208 (2013).
Degenhardt, E. K. et al. Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate Alzheimer dementia. Psychosomatics 57 , 208–216 (2016).
Robinson, J. L. et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141 , 2181–2193 (2018).
Prince, M. et al. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res. Ther. 8 , 23 (2016).
Röhr, S. et al. Estimating prevalence of subjective cognitive decline in and across international cohort studies of aging: a COSMIC study. Alzheimers Res. Ther. 12 , 167 (2020).
Petersen, R. C. et al. Practice guideline update summary: mild cognitive impairment: report of the guideline development, dissemination, and implementation subcommittee of the american academy of neurology. Neurology 90 , 126–135 (2018).
Petersen, R. C. et al. Prevalence of mild cognitive impairment is higher in men than in women. The Mayo Clinic Study of Aging. Neurology 75 , 889–897 (2010).
Mielke, M. M., Vemuri, P. & Rocca, W. A. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin. Epidemiol. 6 , 37–48 (2014).
Thambisetty, M., An, Y. & Tanaka, T. Alzheimer’s disease risk genes and the age-at-onset phenotype. Neurobiol. Aging 34 , 2696.e1–5 (2013).
CAS Google Scholar
Haass, C., Kaether, C., Thinakaran, G. & Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2 , a006270 (2012).
Jonsson, T. et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488 , 96–99 (2012).
van der Lee, S. J. et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 17 , 434–444 (2018).
Bellenguez, C. et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 59 , 220.e1–220.e9 (2017).
Leonenko, G. et al. Polygenic risk and hazard scores for Alzheimer’s disease prediction. Ann. Clin. Transl. Neurol. 6 , 456–465 (2019).
Karch, C. M., Cruchaga, C. & Goate, A. M. Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83 , 11–26 (2014).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51 , 414–430 (2019).
Livingston, G. et al. Dementia prevention, intervention, and care: 2020 report of the Lancet commission. Lancet 396 , 413–446 (2020).
Singh-Manoux, A. et al. Trajectories of depressive symptoms before diagnosis of dementia: a 28-year follow-up study. JAMA Psychiatry 74 , 712–718 (2017).
Gottesman, R. F. et al. Associations Between Midlife Vascular Risk Factors and 25-Year Incident Dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol. 74 , 1246–1254 (2017).
Samieri, C. et al. Association of cardiovascular health level in older age with cognitive decline and incident dementia. JAMA 320 , 657–664 (2018).
Gottesman, R. F. et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317 , 1443–1450 (2017).
Vemuri, P. et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain 138 , 761–771 (2015).
Golde, T. E., DeKosky, S. T. & Galasko, D. Alzheimer’s disease: the right drug, the right time. Science 362 , 1250–1251 (2018).
Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18 , 794–799 (2015).
Zlokovic, B. V. et al. Vascular contributions to cognitive impairment and dementia (VCID): a report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimers Dement. 16 , 1714–1733 (2020).
Arnold, S. E., Hyman, B. T., Flory, J., Damasio, A. R. & Van Hoesen, G. W. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1 , 103–116 (1991). Graphic demonstration of the different regional distributions of Aβ and tau at autopsy.
Montine, T. J. et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123 , 1–11 (2012).
Haass, C. & Selkoe, D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8 , 101–112 (2007).
Holtzman, D. M., Morris, J. C. & Goate, A. M. Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3 , 77sr71 (2011).
Thinakaran, G. & Koo, E. H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283 , 29615–29619 (2008).
Haass, C. & Willem, M. Secreted APP modulates synaptic activity: a novel target for therapeutic intervention? Neuron 101 , 557–559 (2019).
Cirrito, J. R. et al. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48 , 913–922 (2005).
Kang, J. E. et al. Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326 , 1005–1007 (2009).
Boespflug, E. L. & Iliff, J. J. The emerging relationship between interstitial fluid-cerebrospinal fluid exchange, amyloid-β, and sleep. Biol. Psychiatry 83 , 328–336 (2018).
Spires-Jones, T. L. & Hyman, B. T. The intersection of amyloid β and tau at synapses in Alzheimer’s disease. Neuron 82 , 756–771 (2014).
Benarroch, E. E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: emerging mechanisms. Neurology 91 , 125–132 (2018).
Rice, H. C. et al. Secreted amyloid-β precursor protein functions as a GABA(B)R1a ligand to modulate synaptic transmission. Science 363 , eaao4827 (2019).
Müller, U. C., Deller, T. & Korte, M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 18 , 281–298 (2017).
Small, S. A. & Petsko, G. A. Endosomal recycling reconciles the Alzheimer’s disease paradox. Sci. Transl. Med. 12 , eabb1717 (2020).
Gallardo, G. & Holtzman, D. M. Amyloid-β and tau at the crossroads of Alzheimer’s disease. Adv. Exp. Med. Biol. 1184 , 187–203 (2019).
Pooler, A. M., Noble, W. & Hanger, D. P. A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology 76 (Pt A), 1–8 (2014).
Eftekharzadeh, B. et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99 , 925–940.e7 (2018).
Kent, S. A., Spires-Jones, T. L. & Durrant, C. S. The physiological roles of tau and Aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 140 , 417–447 (2020).
Yamada, K. et al. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211 , 387–393 (2014).
Wu, J. W. et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19 , 1085–1092 (2016).
de Calignon, A. et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73 , 685–697 (2012).
Dujardin, S. et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 26 , 1256–1263 (2020). A clinical-neuropathological-molecular demonstration of how post-translational modifications of tau might play a role in the rate of clinical progression.
Shi, Y. et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549 , 523–527 (2017).
Gratuze, M. et al. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Invest. 130 , 4954–4968 (2020).
Shi, Y. et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 216 , 2546–2561 (2019).
Braak, H., Thal, D. R., Ghebremedhin, E. & Del Tredici, K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70 , 960–969 (2011).
Braak, H. & Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82 , 239–259 (1991).
Delacourte, A. et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 52 , 1158–1165 (1999).
Price, J. L. & Morris, J. C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 45 , 358–368 (1999).
Delacourte, A. et al. Tau aggregation in the hippocampal formation: an ageing or a pathological process? Exp. Gerontol. 37 , 1291–1296 (2002).
Crary, J. F. et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128 , 755–766 (2014).
Jack, C. R. et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain 142 , 3230–3242 (2019).
Hanseeuw, B. J. et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 76 , 915–924 (2019).
Ossenkoppele, R. et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139 , 1551–1567 (2016).
Graff-Radford, J. et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 20 , 222–234 (2021).
Kim, J., Basak, J. M. & Holtzman, D. M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 63 , 287–303 (2009).
Huynh, T. V., Davis, A. A., Ulrich, J. D., Holtzman, D. M. & Apolipoprotein, E. and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res. 58 , 824–836 (2017).
Reiman, E. M. et al. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl Acad. Sci. USA 106 , 6820–6825 (2009).
Jack, C. R. Jr. et al. Age, sex, and APOE e4 effects on memory, brain structure, and β-amyloid across the adult life span. JAMA Neurol. 72 , 511–519 (2015).
van der Kant, R., Goldstein, L. S. B. & Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21 , 21–35 (2020).
Small, S. A. & Duff, K. Linking Aβ and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron 60 , 534–542 (2008).
Busche, M. A. & Hyman, B. T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 23 , 1183–1193 (2020).
Kurt, M. A., Davies, D. C., Kidd, M., Duff, K. & Howlett, D. R. Hyperphosphorylated tau and paired helical filament-like structures in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Neurobiol. Dis. 14 , 89–97 (2003).
Le, R. et al. Plaque-induced abnormalities in neurite geometry in transgenic models of Alzheimer disease: implications for neural system disruption. J. Neuropathol. Exp. Neurol. 60 , 753–758 (2001).
Leyns, C. E. G. et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 22 , 1217–1222 (2019).
Small, S. A., Simoes-Spassov, S., Mayeux, R. & Petsko, G. A. Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer’s disease. Trends Neurosci. 40 , 592–602 (2017).
Nixon, R. A. & Yang, D. S. Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol. Dis. 43 , 38–45 (2011).
Palop, J. J. & Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17 , 777–792 (2016).
Terry, R. D. et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30 , 572–580 (1991).
DeKosky, S. T. & Scheff, S. W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27 , 457–464 (1990). A real-time demonstration of a close correspondence between synapse loss and degree of cognitive impairment in brain biopsies from patients with AD.
Spires, T. L. et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 25 , 7278–7287 (2005).
Arbel-Ornath, M. et al. Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 12 , 27 (2017).
Zhou, L. et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 8 , 15295 (2017).
Masliah, E., Hansen, L., Albright, T., Mallory, M. & Terry, R. D. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 81 , 428–433 (1991).
Henstridge, C. M. et al. Post-mortem brain analyses of the Lothian Birth Cohort 1936: extending lifetime cognitive and brain phenotyping to the level of the synapse. Acta Neuropathol. Commun. 3 , 53 (2015).
Lleo, A. et al. Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s Disease cerebrospinal fluid. Mol. Cell Proteom. 18 , 546–560 (2019).
DeVos, S. L. et al. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front. Neurosci. 12 , 267 (2018).
Yoshiyama, Y. et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53 , 337–351 (2007).
Keskin, A. D. et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc. Natl Acad. Sci. USA 114 , 8631–8636 (2017).
Busche, M. A. et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321 , 1686–1689 (2008).
Kuchibhotla, K. V. et al. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59 , 214–225 (2008).
Menkes-Caspi, N. et al. Pathological tau disrupts ongoing network activity. Neuron 85 , 959–966 (2015).
Busche, M. A. et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 22 , 57–64 (2019).
Anderson, K. J., Scheff, S. W. & DeKosky, S. T. Reactive synaptogenesis in hippocampal area CA1 of aged and young adult rats. J. Comp. Neurol. 252 , 374–384 (1986).
Geddes, J. W. & Cotman, C. W. Plasticity in hippocampal excitatory amino acid receptors in Alzheimer’s disease. Neurosci. Res. 3 , 672–678 (1986).
Hyman, B. T., Kromer, L. J. & Van Hoesen, G. W. Reinnervation of the hippocampal perforant pathway zone in Alzheimer’s disease. Ann. Neurol. 21 , 259–267 (1987).
Liu, L. et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7 , e31302 (2012).
Rauch, J. N. et al. LRP1 is a master regulator of tau uptake and spread. Nature 580 , 381–385 (2020).
Kounnas, M. Z. et al. LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell 82 , 331–340 (1995).
Rebeck, G. W., Reiter, J. S., Strickland, D. K. & Hyman, B. T. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 11 , 575–580 (1993).
Wilton, D. K., Dissing-Olesen, L. & Stevens, B. Neuron-glia signaling in synapse elimination. Annu. Rev. Neurosci. 42 , 107–127 (2019).
Nixon, R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 19 , 983–997 (2013).
Menzies, F. M. et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93 , 1015–1034 (2017).
Nixon, R. A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J. 31 , 2729–2743 (2017).
Shehata, M. et al. Autophagy enhances memory erasure through synaptic destabilization. J. Neurosci. 38 , 3809–3822 (2018).
Glatigny, M. et al. Autophagy is required for memory formation and reverses age-related memory decline. Curr. Biol. 29 , 435–448.e8 (2019).
Cataldo, A. M. et al. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157 , 277–286 (2000).
Van Acker, Z. P., Bretou, M. & Annaert, W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol. Neurodegener. 14 , 20 (2019).
Suire, C. N. et al. Cathepsin D regulates cerebral Aβ42/40 ratios via differential degradation of Aβ42 and Aβ40. Alzheimers Res. Ther. 12 , 80 (2020).
Kwart, D. et al. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 104 , 256–270.e5 (2019). Evidence from induced pluripotent stem cells on the critical role of endosomal dysregulation in the pathogenesis of Aβ and tau-induced disease.
Pensalfini, A. et al. Endosomal dysfunction induced by directly over-activating Rab5 recapitulates prodromal and neurodegenerative features of Alzheimer’s disease. Cell Rep. 33 , 108420 (2020).
Lauritzen, I. et al. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 132 , 257–276 (2016).
Lee, J. H. et al. Presenilin 1 maintains lysosomal Ca 2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12 , 1430–1444 (2015).
Lee, J. H. et al. β2-adrenergic agonists rescue lysosome acidification and function in PSEN1 deficiency by reversing defective ER-to-lysosome delivery of ClC-7. J. Mol. Biol. 432 , 2633–2650 (2020).
Morel, N. & Poea-Guyon, S. The membrane domain of vacuolar H + ATPase: a crucial player in neurotransmitter exocytotic release. Cell. Mol. Life Sci. 72 , 2561–2573 (2015).
Higashida, H., Yokoyama, S., Tsuji, C. & Muramatsu, S. I. Neurotransmitter release: vacuolar ATPase V0 sector c-subunits in possible gene or cell therapies for Parkinson’s, Alzheimer’s, and psychiatric diseases. J. Physiol. Sci. 67 , 11–17 (2017).
El Far, O. & Seagar, M. A role for V-ATPase subunits in synaptic vesicle fusion? J. Neurochem. 117 , 603–612 (2011).
Peng, K. Y. et al. Apolipoprotein E4 genotype compromises brain exosome production. Brain 142 , 163–175 (2019).
Liu, R. Q. et al. Membrane localization of β-amyloid 1-42 in lysosomes: a possible mechanism for lysosome labilization. J. Biol. Chem. 285 , 19986–19996 (2010).
Morishita, H. & Mizushima, N. Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol. 35 , 453–475 (2019).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176 , 11–42 (2019).
Bordi, M. et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12 , 2467–2483 (2016).
Moreira, P. I., Carvalho, C., Zhu, X., Smith, M. A. & Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 1802 , 2–10 (2010).
Nixon, R. A. The aging lysosome: an essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 1868 , 140443 (2020).
Whyte, L. S., Lau, A. A., Hemsley, K. M., Hopwood, J. J. & Sargeant, T. J. Endo-lysosomal and autophagic dysfunction: a driving factor in Alzheimer’s disease? J. Neurochem. 140 , 703–717 (2017).
Pensalfini, A. et al. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol. Dis. 71 , 53–61 (2014).
Adalbert, R. et al. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 132 , 402–416 (2009).
Nixon, R. A. & Yang, D. S. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb. Perspect. Biol. 4 , a008839 (2012).
Nakanishi, H. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regenerat. Res. 15 , 25–29 (2020).
Lowry, J. R. & Klegeris, A. Emerging roles of microglial cathepsins in neurodegenerative disease. Brain Res. Bull. 139 , 144–156 (2018).
Sarlus, H. & Heneka, M. T. Microglia in Alzheimer’s disease. J. Clin. Invest. 127 , 3240–3249 (2017).
Deretic, V., Saitoh, T. & Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13 , 722–737 (2013).
Cho, M. H. et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10 , 1761–1775 (2014).
Jay, T. R. et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37 , 637–647 (2017).
Lewcock, J. W. et al. Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron 108 , 801–821 (2020).
Kim, H. J. et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 22 , 1576–1584 (2017).
Saido, T. & Leissring, M. A. Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2 , a006379 (2012).
Rasmussen, M. K., Mestre, H. & Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 17 , 1016–1024 (2018).
Buckner, R. L., Andrews-Hanna, J. R. & Schacter, D. L. The brain’s default network: anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci. 1124 , 1–38 (2008). Demonstration of the relationship between the default mode network and amyloid accumulation.
Greicius, M. D., Srivastava, G., Reiss, A. L. & Menon, V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc. Natl Acad. Sci. USA 101 , 4637–4642 (2004).
Buckner, R. L. et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J. Neurosci. 29 , 1860–1873 (2009).
Agosta, F. et al. Resting state fMRI in Alzheimer’s disease: beyond the default mode network. Neurobiol. Aging 33 , 1564–1578 (2012).
Whitwell, J. L. et al. Working memory and language network dysfunctions in logopenic aphasia: a task-free fMRI comparison with Alzheimer’s dementia. Neurobiol. Aging 36 , 1245–1252 (2015).
Townley, R. A. et al. Progressive dysexecutive syndrome due to Alzheimer’s disease: a description of 55 cases and comparison to other phenotypes. Brain Commun. 2 , fcaa068 (2020).
Franzmeier, N. et al. Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat. Commun. 11 , 347 (2020).
Sintini, I. et al. Tau and amyloid relationships with resting-state functional connectivity in atypical Alzheimer’s disease. Cereb. Cortex https://doi.org/10.1093/cercor/bhaa319 (2020).
Article PubMed Central Google Scholar
Jones, D. T. et al. Tau, amyloid, and cascading network failure across the Alzheimer’s disease spectrum. Cortex 97 , 143–159 (2017).
Sepulcre, J. et al. Hierarchical organization of tau and amyloid deposits in the cerebral cortex. JAMA Neurol. 74 , 813–820 (2017).
Jack, C. R. Jr. et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology 81 , 1732–1740 (2013).
Protas, H. D. et al. Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late-middle-aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA Neurol. 70 , 320–325 (2013).
Franzmeier, N. et al. Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer’s disease. Sci. Adv. 6 , eabd1327 (2020). Analysis showing how regional tau expansion follows connectivity patterns.
D’Onofrio, G. et al. Neuropsychiatric symptoms and functional status in Alzheimer’s disease and vascular dementia patients. Curr. Alzheimer Res. 9 , 759–771 (2012).
Lehmann, M. et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain 136 , 844–858 (2013).
Crutch, S. J. et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 13 , 870–884 (2017).
Gorno-Tempini, M. L. et al. The logopenic/phonological variant of primary progressive aphasia. Neurology 71 , 1227–1234 (2008).
Bergeron, D. et al. Prevalence of amyloid-β pathology in distinct variants of primary progressive aphasia. Ann. Neurol. 84 , 729–740 (2018).
Ossenkoppele, R. et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 138 , 2732–2749 (2015).
Murray, M. E. et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 10 , 785–796 (2011).
Clarfield, A. M. The decreasing prevalence of reversible dementias: an updated meta-analysis. Arch. Intern. Med. 163 , 2219–2229 (2003).
Beach, T. G., Monsell, S. E., Phillips, L. E. & Kukull, W. Accuracy of the clinical diagnosis of Alzheimer disease at national institute on aging Alzheimer disease centers, 2005-2010. J. Neuropathol. Exp. Neurol. 71 , 266–273 (2012).
McKeith, I. G. et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 89 , 88–100 (2017).
Rascovsky, K. et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134 , 2456–2477 (2011).
Gorno-Tempini, M. L. et al. Classification of primary progressive aphasia and its variants. Neurology 76 , 1006–1014 (2011).
Botha, H. et al. FDG-PET in tau-negative amnestic dementia resembles that of autopsy-proven hippocampal sclerosis. Brain 141 , 1201–1217 (2018).
Petersen, R. C. & Yaffe, K. Issues and questions surrounding screening for cognitive impairment in older patients. JAMA 323 , 722–724 (2020).
Patnode, C. D. et al. Screening for cognitive impairment in older adults: updated evidence report and systematic review for the US Preventive Services TaskFforce. JAMA 323 , 764–785 (2020).
Jack, C. R. Jr. et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14 , 535–562 (2018).
Chetelat, G. et al. Amyloid-PET and 18 F-FDG-PET in the diagnostic investigation of Alzheimer”s disease and other dementias. Lancet Neurol. 19 , 951–962 (2020).
Klunk, W. E. et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 55 , 306–319 (2004).
Jack, C. R. J. et al. Introduction to revised criteria for the diagnosis of Alzheimer’s disease: National Institute on Aging and the Alzheimer Association Workgroups. Alzheimer’s Dement. 7 , 257–262 (2011).
Knopman, D. S. et al. Evolution of neurodegeneration-imaging biomarkers from clinically normal to dementia in the Alzheimer disease spectrum. Neurobiol. Aging 46 , 32–42 (2016).
Schröder, J. & Pantel, J. Neuroimaging of hippocampal atrophy in early recognition of Alzheimer’s disease–a critical appraisal after two decades of research. Psychiatry Res. Neuroimaging 247 , 71–78 (2016).
Petersen, C. et al. Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol. 138 , 597–612 (2019).
Ridha, B. H. et al. Tracking atrophy progression in familial Alzheimer’s disease: a serial MRI study. Lancet Neurol. 5 , 828–834 (2006).
Greenberg, S. M. et al. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat. Rev. Neurol. 16 , 30–42 (2020).
Graff-Radford, J. et al. Cerebral microbleeds: prevalence and relationship to amyloid burden. Neurology 92 , e253–e262 (2019).
Li, X. et al. The significant effects of cerebral microbleeds on cognitive dysfunction: an updated meta-analysis. PLoS ONE 12 , e0185145 (2017).
Laforce, R. Jr. et al. Parallel ICA of FDG-PET and PiB-PET in three conditions with underlying Alzheimer’s pathology. Neuroimage Clin. 4 , 508–516 (2014).
Caroli, A. et al. Mild cognitive impairment with suspected nonamyloid pathology (SNAP): prediction of progression. Neurology 84 , 508–515 (2015).
Iaccarino, L., Sala, A. & Perani, D. Predicting long-term clinical stability in amyloid-positive subjects by FDG-PET. Ann. Clin. Transl. Neurol. 6 , 1113–1120 (2019).
Villemagne, V. L., Doré, V., Burnham, S. C., Masters, C. L. & Rowe, C. C. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 14 , 225–236 (2018).
Clark, C. M. et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 11 , 669–678 (2012).
Murray, M. E. et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain 138 , 1370–1381 (2015).
Klunk, W. E. et al. The centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 11 , 1–15.e1–4 (2015).
Landau, S. M. et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid. Ann. Neurol. 74 , 826–836 (2013).
Leuzy, A. et al. Pittsburgh compound B imaging and cerebrospinal fluid amyloid-β in a multicentre European memory clinic study. Brain 139 , 2540–2553 (2016).
Grothe, M. J. et al. In vivo staging of regional amyloid deposition. Neurology 89 , 2031–2038 (2017).
Jagust, W. J. & Landau, S. M. Temporal dynamics of β-amyloid accumulation in aging and Alzheimer’s disease. Neurology 96 , e1347–e1357 (2021).
Jack, C. R. Jr. et al. Prevalence of biologically vs clinically defined Alzheimer spectrum entities using THE National Institute on Aging-Alzheimer’s association research framework. JAMA Neurol. 76 , 1174–1183 (2019).
PubMed Central Google Scholar
Johnson, K. A. et al. Appropriate use criteria for amyloid PET: a report of the amyloid imaging task force, the society of nuclear medicine and molecular imaging, and the Alzheimer’s association. J. Nucl. Med. 54 , 476–490 (2013).
Rabinovici, G. D. et al. Association of amyloid positron emission tomography with subsequent change in clinical management among medicare beneficiaries with mild cognitive impairment or dementia. JAMA 321 , 1286–1294 (2019). A very large pragmatic trial of the value of Aβ imaging in clinical dementia practice.
de Wilde, A. et al. Association of amyloid positron emission tomography with changes in diagnosis and patient treatment in an unselected memory clinic cohort: the ABIDE project. JAMA Neurol. 75 , 1062–1070 (2018).
Jeong, H. J. et al. [ 18 F]THK5351 PET imaging in patients with mild cognitive impairment. J. Clin. Neurol. 16 , 202–214 (2020).
Pascoal, T. A. et al. 18 F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 143 , 2818–2830 (2020).
Leuzy, A. et al. Diagnostic performance of RO948 F 18 tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol. 77 , 955–965 (2020).
Ossenkoppele, R. et al. Discriminative accuracy of [ 18 F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 320 , 1151–1162 (2018). Demonstration of the remarkable specificity of an elevated tau PET signal outside of the medial temporal lobe for persons with elevated Aβ.
Aschenbrenner, A. J., Gordon, B. A., Benzinger, T. L. S., Morris, J. C. & Hassenstab, J. J. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology 91 , e859–e866 (2018).
Brier, M. R. et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 8 , 338ra366 (2016).
Harrison, T. M. et al. Longitudinal tau accumulation and atrophy in aging and Alzheimer disease. Ann. Neurol. 85 , 229–240 (2019).
Lu, M. et al. Aggregated tau measured by visual interpretation of flortaucipir positron emission tomography and the associated risk of clinical progression of mild cognitive impairment and Alzheimer disease: results from 2 phase III clinical trials. JAMA Neurol. 78 , 445–453 (2021).
Lowe, V. J. et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141 , 271–287 (2018).
Pontecorvo, M. J. et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140 , 748–763 (2017).
Shaw, L. M. et al. Appropriate use criteria for lumbar puncture and cerebrospinal fluid testing in the diagnosis of Alzheimer’s disease. Alzheimers Dement. 14 , 1505–1521 (2018).
Molinuevo, J. L. et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 136 , 821–853 (2018).
Hansson, O. et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 1 , 30029–30023 (2018).
Hansson, O. et al. Prediction of Alzheimer’s disease using the CSF Aβ42/Aβ40 ratio in patients with mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 23 , 316–320 (2007).
Wiltfang, J. et al. Amyloid β peptide ratio 42/40 but not Aβ42 correlates with phospho-Tau in patients with low- and high-CSF Aβ40 load. J. Neurochem. 101 , 1053–1059 (2007).
Mattsson, N. et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302 , 385–393 (2009).
Skillbäck, T. et al. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 138 , 2716–2731 (2015).
Buerger, K. et al. No correlation between CSF tau protein phosphorylated at threonine 181 with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 130 , e82 (2007).
Seppälä, T. T. et al. CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology 78 , 1568–1575 (2012).
Barthélemy, N. R. et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 26 , 398–407 (2020).
Janelidze, S. et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 11 , 1683 (2020).
Kern, S. et al. Association of cerebrospinal fluid neurofilament light protein with risk of mild cognitive impairment among individuals without cognitive impairment. JAMA Neurol. 76 , 187–193 (2019).
Zetterberg, H. & Bendlin, B. B. Biomarkers for Alzheimer’s disease-preparing for a new era of disease-modifying therapies. Mol. Psychiatry 26 , 296–308 (2020).
Tarawneh, R. et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 73 , 561–571 (2016).
Schindler, S. E. et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 93 , e1647–e1659 (2019).
Karikari, T. K. et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 19 , 422–433 (2020).
Palmqvist, S. et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 324 , 772–781 (2020).
de Wolf, F. et al. Plasma tau, neurofilament light chain and amyloid-β levels and risk of dementia; a population-based cohort study. Brain 143 , 1220–1232 (2020).
Ashton, N. J. et al. An update on blood-based biomarkers for non-Alzheimer neurodegenerative disorders. Nat. Rev. Neurol. 16 , 265–284 (2020).
Wattmo, C. & Wallin A, K. Early- versus late-onset Alzheimer’s disease in clinical practice: cognitive and global outcomes over 3 years. Alzheimers Res. Ther. 9 , 70 (2017).
Wilson, R. S. et al. Cognitive decline in incident Alzheimer’s disease in a community population. Neurology 74 , 951–955 (2010).
van Harten, A. C. et al. Subjective cognitive decline and risk of MCI: the Mayo Clinic Study of Aging. Neurology 91 , e300–e312 (2018).
Kryscio, R. J. et al. Self-reported memory complaints: implications from a longitudinal cohort with autopsies. Neurology 83 , 1359–1365 (2014).
Stewart, R. et al. Longitudinal neuroimaging correlates of subjective memory impairment: 4-year prospective community study. Br. J. Psychiatry 198 , 199–205 (2011).
Amariglio, R. E. et al. Subjective cognitive complaints and amyloid burden in cognitively normal older individuals. Neuropsychologia 50 , 2880–2886 (2012).
Petersen, R. C. et al. Randomized controlled trials in mild cognitive impairment: sources of variability. Neurology 88 , 1751–1758 (2017).
Roberts, R. O. et al. Prevalence and outcomes of amyloid positivity among persons without dementia in a longitudinal, population-based setting. JAMA Neurol. 75 , 970–979 (2018).
Vos, S. J. et al. Prevalence and prognosis of Alzheimer’s disease at the mild cognitive impairment stage. Brain 138 , 1327–1338 (2015).
Jack, C. R. Jr. et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA 321 , 2316–2325 (2019).
Knopman, D. S. et al. Entorhinal cortex tau, amyloid-β, cortical thickness and memory performance in non-demented subjects. Brain 142 , 1148–1160 (2019).
Vos, S. J. et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 12 , 957–965 (2013).
Serrano-Pozo, A., Qian, J., Monsell, S. E., Betensky, R. A. & Hyman, B. T. APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann. Neurol. 77 , 917–929 (2015).
Craft, S. et al. Accelerated decline in apolipoprotein E-epsilon4 homozygotes with Alzheimer’s disease. Neurology 51 , 149–153 (1998).
Butler, M. et al. Over-the-counter supplement interventions to prevent cognitive decline, mild cognitive impairment, and clinical Alzheimer-type dementia: a systematic review. Ann. Intern. Med. 168 , 52–62 (2018).
Fink, H. A. et al. Pharmacologic interventions to prevent cognitive decline, mild cognitive impairment, and clinical Alzheimer-type dementia: a systematic review. Ann. Intern. Med. 168 , 39–51 (2018).
Brasure, M. et al. Physical activity interventions in preventing cognitive decline and Alzheimer-type dementia: a systematic review. Ann. Intern. Med. 168 , 30–38 (2018).
Kane, R. L. et al. in Interventions to Prevent Age-Related Cognitive Decline, Mild Cognitive Impairment, and Clinical Alzheimer’s-Type Dementia (Agency for Healthcare Research and Quality, 2017).
Ngandu, T. et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet 385 , 2255–2263 (2015). A first demonstration of non-pharmacological means of delaying cognitive decline in elderly persons at risk for dementia.
Debette, S. et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology 77 , 461–468 (2011).
Williamson, J. D. et al. Effect of intensive vs standard blood pressure control on probable dementia: a randomized clinical trial. JAMA 321 , 553–561 (2019).
Reuben, D. B. et al. D-CARE: the dementia care study: design of a pragmatic trial of the effectiveness and cost effectiveness of health system-based versus community-based dementia care versus usual dementia care. J. Am. Geriatr. Soc. 68 , 2492–2499 (2020).
Fisk, J. D., Beattie, B. L., Donnelly, M., Byszewski, A. & Molnar, F. J. Disclosure of the diagnosis of dementia. Alzheimers Dement. 3 , 404–410 (2007).
Alpinar-Sencan, Z. & Schicktanz, S. Addressing ethical challenges of disclosure in dementia prediction: limitations of current guidelines and suggestions to proceed. BMC Med. Ethics 21 , 33 (2020).
Amieva, H. et al. Group and individual cognitive therapies in Alzheimer’s disease: the ETNA3 randomized trial. Int. Psychogeriatr. 28 , 707–717 (2016).
Tricco, A. C. et al. Comparisons of interventions for preventing falls in older adults: a systematic review and meta-analysis. JAMA 318 , 1687–1699 (2017).
Mohs, R. C. et al. A 1-year, placebo-controlled preservation of function survival study of donepezil in AD patients. Neurology 57 , 481–488 (2001).
Panza, F., Lozupone, M., Logroscino, G. & Imbimbo, B. P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 15 , 73–88 (2019).
Rogers, M. B. https://www.alzforum.org/news/research-news/aducanumab-still-needs-prove-itself-researchers-say (2020).
Mintun, M. A. et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med . https://doi.org/10.1056/NEJMoa2100708 (2021).
Shugart, J. https://www.alzforum.org/news/conference-coverage/banish-av-ban2401-antibody-makes-its-move-phase-3-program (2020).
VandeVrede, L., Boxer, A. L. & Polydoro, M. Targeting tau: clinical trials and novel therapeutic approaches. Neurosci. Lett. 731 , 134919 (2020).
Rios-Romenets, S. et al. Baseline demographic, clinical, and cognitive characteristics of the Alzheimer’s prevention initiative (API) autosomal-dominant Alzheimer’s disease Colombia trial. Alzheimers Dement 16 , 1023–1030 (2020).
Insel, P. S., Donohue, M. C., Sperling, R., Hansson, O. & Mattsson-Carlgren, N. The A4 study: β-amyloid and cognition in 4432 cognitively unimpaired adults. Ann. Clin. Transl. Neurol. 7 , 776–785 (2020).
Ballard, C. et al. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol. 17 , 213–222 (2018).
Jennings, L. A. et al. Patient and caregiver goals for dementia care. Qual. Life Res. 26 , 685–693 (2017).
Bannon, S. et al. In it together: a qualitative meta-synthesis of common and unique psychosocial stressors and adaptive coping strategies of persons with young-onset dementia and their caregivers. Gerontologist https://doi.org/10.1093/geront/gnaa169 (2020).
Moon, H. & Adams, K. B. The effectiveness of dyadic interventions for people with dementia and their caregivers. Dementia 12 , 821–839 (2013).
van Charante, E. P. et al. Effectiveness of a 6-year multidomain vascular care intervention to prevent dementia (preDIVA): a cluster-randomised controlled trial. Lancet 388 , 797–805 (2016).
Andrieu, S. et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): a randomised, placebo-controlled trial. Lancet Neurol. 16 , 377–389 (2017).
Sink, K. M. et al. Effect of a 24-month physical activity intervention vs health education on cognitive outcomes in sedentary older adults: the life randomized trial. JAMA 314 , 781–790 (2015).
De Jager, P. L., Yang, H. S. & Bennett, D. A. Deconstructing and targeting the genomic architecture of human neurodegeneration. Nat. Neurosci. 21 , 1310–1317 (2018).
Rehiman, S. H. et al. Proteomics as a reliable approach for discovery of blood-based Alzheimer’s disease biomarkers: a systematic review and meta-analysis. Ageing Res. Rev. 60 , 101066 (2020).
Mahajan, U. V. et al. Dysregulation of multiple metabolic networks related to brain transmethylation and polyamine pathways in Alzheimer disease: a targeted metabolomic and transcriptomic study. PLoS Med. 17 , e1003012 (2020).
Dawson, T. M., Golde, T. E. & Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 21 , 1370–1379 (2018).
van der Kant, R. et al. Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 24 , 363–375.e9 (2019).
American Psychiatric Association. DSM-5: Diagnostic and Statistical Manual of Mental Disorders 5th ed. (American Psychiatric Association, 2013).
Albert, M. et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging– Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 7 , 270–279 (2011).
Dubois, B. et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 13 , 614–629 (2014).
Thal, D. R., Rub, U., Orantes, M. & Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58 , 1791–1800 (2002).
Download references
Acknowledgements
The authors acknowledge research support from NIH (D.S.K. and R.C.P, P30 AG062677 and U01 AG006786; B.T.H., P30AG062421; R.A.N. P01 AG017617 and R01 AG062376).
Author information
Authors and affiliations.
Department of Neurology, Mayo Clinic, Rochester, MN, USA
David S. Knopman, Ronald C. Petersen & David T. Jones
Inserm U1219 Bordeaux Population Health Center, University of Bordeaux, Bordeaux, France
Helene Amieva
Normandie Univ, UNICAEN, INSERM, U1237, PhIND “Physiopathology and Imaging of Neurological Disorders”, Institut Blood and Brain @ Caen-Normandie, Cyceron, Caen, France
Gäel Chételat
Department of Neurology, Washington University School of Medicine, St. Louis, MO, USA
David M. Holtzman
Department of Neurology, Massachusetts General Hospital, Boston, MA, USA
Bradley T. Hyman
Departments of Psychiatry and Cell Biology, New York University Langone Medical Center, New York University, New York, NY, USA
Ralph A. Nixon
NYU Neuroscience Institute, New York University Langone Medical Center, New York University, New York, NY, USA
You can also search for this author in PubMed Google Scholar
Contributions
Introduction (D.S.K.); Epidemiology (H.A.); Mechanisms/pathophysiology (D.T.J., R.A.N., B.T.H. and D.M.H.); Diagnosis, screening and prevention (G.C., R.C.P. and D.S.K.); Management (R.C.P. and D.S.K.); Quality of life (D.S.K.); Outlook (D.S.K.); Overview of Primer (D.S.K.).
Corresponding author
Correspondence to David S. Knopman .
Ethics declarations
Competing interests.
D.S.K. served on a Data Safety Monitoring Board for the DIAN study. He serves on a Data Safety Monitoring Board for a tau therapeutic for Biogen but receives no personal compensation. He is a site investigator in a Biogen aducanumab trial. He is an investigator in a clinical trial sponsored by Lilly Pharmaceuticals and the University of Southern California. He serves as a consultant for Samus Therapeutics, Third Rock, Roche and Alzeca Biosciences but receives no personal compensation. He receives research support from the NIH. G.C. serves on the Scientific Advisory Board of the Fondation Vaincre Alzheimer but receives no personal compensation. She receives personal fees from Fondation d’entreprise MMA des Entrepreneurs du Futur and from Fondation Alzheimer as she serves in the Operational Committee. She receives research support from European Union Horizon 2020 research and innovation programme (grant agreement number 667696), Inserm, Fondation d’entreprise MMA des Entrepreneurs du Futur, Fondation Alzheimer, Programme Hospitalier de Recherche Clinique, Région Normandie, Association France Alzheimer et maladies apparentées and Fondation Vaincre Alzheimer. R.C.P. is a consultant for Biogen, Inc., Roche, Inc., Merck, Inc., Genentech Inc. and Eisai, Inc., has given educational lectures for GE Healthcare, receives publishing royalties from Mild Cognitive Impairment (Oxford University Press, 2003), UpToDate, and receives research support from the NIH. B.T.H. has a family member who works at Novartis and owns stock in Novartis; he serves on the SAB of Dewpoint and owns stock. He serves on a scientific advisory board or is a consultant for Biogen, Novartis, Cell Signalling, the US Dept of Justice, Takeda, Vigil, W20 group and Seer. His laboratory is supported by sponsored research agreements with Abbvie, F Prim, and research grants from the National Institutes of Health, Cure Alzheimer’s Fund, Tau Consortium, Brightfocus and the JPB Foundation. H.A. serves on the Scientific Advisory Board of the Observatoire des Mémoires but receives no personal compensation. She receives research support from Spoelberch Foundation, Association France Alzheimer et maladies apparentées, the Regional Health Agency of Aquitaine and National Research Agency. D.M.H. reports being a Co-founder for C2N Diagnostics LLC and participating in scientific advisory boards/consulting for Genentech, C2N Diagnostics, Denali, Merck and Idorsia. He is an inventor on patents licensed by Washington University to C2N Diagnostics on the therapeutic use of anti-tau antibodies (this anti-tau antibody programme is licensed to Abbvie) and to Eli Lilly on the therapeutic use of an anti-amyloid-β antibody. His laboratory receives research grants from the National Institutes of Health, Cure Alzheimer’s Fund, Tau Consortium, the JPB Foundation, Good Ventures, Centene, BrightFocus and C2N Diagnostics. All other authors declare no competing interests.
Additional information
Peer review information.
Nature Reviews Disease Primers thanks K. Iqbal, D. Selkoe, H. Soininen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information, rights and permissions.
Reprints and permissions
About this article
Cite this article.
Knopman, D.S., Amieva, H., Petersen, R.C. et al. Alzheimer disease. Nat Rev Dis Primers 7 , 33 (2021). https://doi.org/10.1038/s41572-021-00269-y
Download citation
Accepted : 09 April 2021
Published : 13 May 2021
DOI : https://doi.org/10.1038/s41572-021-00269-y
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Variation of the essential oil components of citrus aurantium leaves upon using different distillation techniques and evaluation of their antioxidant, antidiabetic, and neuroprotective effect against alzheimer’s disease.
- Esraa A. Elhawary
- Nilofar Nilofar
- Omayma A. Eldahshan
BMC Complementary Medicine and Therapies (2024)
Gut microbiota-host lipid crosstalk in Alzheimer’s disease: implications for disease progression and therapeutics
- Ling-Ling Yang
- Xiu-Qing Yao
Molecular Neurodegeneration (2024)
Loss of GDE2 leads to complex behavioral changes including memory impairment
- Daniel Daudelin
- Anna Westerhaus
- Shanthini Sockanathan
Behavioral and Brain Functions (2024)
Profiles of subgingival microbiomes and gingival crevicular metabolic signatures in patients with amnestic mild cognitive impairment and Alzheimer’s disease
- Zhongchen Song
Alzheimer's Research & Therapy (2024)
Early onset diagnosis in Alzheimer’s disease patients via amyloid-β oligomers-sensing probe in cerebrospinal fluid
- Kyeonghwan Kim
- Jong Seung Kim
Nature Communications (2024)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Translational Research newsletter — top stories in biotechnology, drug discovery and pharma.

Call our 24 hours, seven days a week helpline at 800.272.3900
- Professionals

- Younger/Early-Onset Alzheimer's
- Is Alzheimer's Genetic?
- Women and Alzheimer's
- Creutzfeldt-Jakob Disease
- Dementia with Lewy Bodies
- Down Syndrome & Alzheimer's
- Frontotemporal Dementia
- Huntington's Disease
- Mixed Dementia
- Normal Pressure Hydrocephalus
- Posterior Cortical Atrophy
- Parkinson's Disease Dementia
- Vascular Dementia
- Korsakoff Syndrome
- Traumatic Brain Injury (TBI)
- Know the 10 Signs
- Difference Between Alzheimer's & Dementia
- 10 Steps to Approach Memory Concerns in Others
- Medical Tests for Diagnosing Alzheimer's
- Why Get Checked?
- Visiting Your Doctor
- Life After Diagnosis
- Stages of Alzheimer's
- Earlier Diagnosis
- Part the Cloud
- Research Momentum
- Our Commitment to Research
- TrialMatch: Find a Clinical Trial
- What Are Clinical Trials?
- How Clinical Trials Work
- When Clinical Trials End
- Why Participate?
- Talk to Your Doctor
- Clinical Trials: Myths vs. Facts
- Can Alzheimer's Disease Be Prevented?
- Brain Donation
- Navigating Treatment Options
- Lecanemab Approved for Treatment of Early Alzheimer's Disease
- Aducanumab Discontinued as Alzheimer's Treatment
- Medicare Treatment Coverage
- Donanemab for Treatment of Early Alzheimer's Disease — News Pending FDA Review
- Questions for Your Doctor
- Medications for Memory, Cognition and Dementia-Related Behaviors
- Treatments for Behavior
- Treatments for Sleep Changes
- Alternative Treatments
- Facts and Figures
- Assessing Symptoms and Seeking Help
- Now is the Best Time to Talk about Alzheimer's Together
- Get Educated
- Just Diagnosed
- Sharing Your Diagnosis
- Changes in Relationships
- If You Live Alone
- Treatments and Research
- Legal Planning
- Financial Planning
- Building a Care Team
- End-of-Life Planning
- Programs and Support
- Overcoming Stigma
- Younger-Onset Alzheimer's
- Taking Care of Yourself
- Reducing Stress
- Tips for Daily Life
- Helping Family and Friends
- Leaving Your Legacy
- Live Well Online Resources
- Make a Difference
- Daily Care Plan
- Communication and Alzheimer's
- Food and Eating
- Art and Music
- Incontinence
- Dressing and Grooming
- Dental Care
- Working With the Doctor
- Medication Safety
- Accepting the Diagnosis
- Early-Stage Caregiving
- Middle-Stage Caregiving
- Late-Stage Caregiving
- Aggression and Anger
- Anxiety and Agitation
- Hallucinations
- Memory Loss and Confusion
- Sleep Issues and Sundowning
- Suspicions and Delusions
- In-Home Care
- Adult Day Centers
- Long-Term Care
- Respite Care
- Hospice Care
- Choosing Care Providers
- Finding a Memory Care-Certified Nursing Home or Assisted Living Community
- Changing Care Providers
- Working with Care Providers
- Creating Your Care Team
- Long-Distance Caregiving
- Community Resource Finder
- Be a Healthy Caregiver
- Caregiver Stress
- Caregiver Stress Check
- Caregiver Depression
- Changes to Your Relationship
- Grief and Loss as Alzheimer's Progresses
- Home Safety
- Dementia and Driving
- Technology 101
- Preparing for Emergencies
- Managing Money Online Program
- Planning for Care Costs
- Paying for Care
- Health Care Appeals for People with Alzheimer's and Other Dementias
- Social Security Disability
- Medicare Part D Benefits
- Tax Deductions and Credits
- Planning Ahead for Legal Matters
- Legal Documents
ALZ Talks Virtual Events
- ALZNavigator™
- Veterans and Dementia
- The Knight Family Dementia Care Coordination Initiative
- Online Tools
- Asian Americans and Pacific Islanders and Alzheimer's
- Native Americans and Alzheimer's
- Black Americans and Alzheimer's
- Hispanic Americans and Alzheimer's
- LGBTQ+ Community Resources for Dementia
- Educational Programs and Dementia Care Resources
- Brain Facts
- 50 Activities
- For Parents and Teachers
- Resolving Family Conflicts
- Holiday Gift Guide for Caregivers and People Living with Dementia
- Trajectory Report
- Resource Lists
- Search Databases
- Publications
- Favorite Links
- 10 Healthy Habits for Your Brain
- Stay Physically Active
- Adopt a Healthy Diet
- Stay Mentally and Socially Active
- Online Community
- Support Groups
Find Your Local Chapter
- Any Given Moment
- New IDEAS Study
- RFI Amyloid PET Depletion Following Treatment
- Bruce T. Lamb, Ph.D., Chair
- Christopher van Dyck, M.D.
- Cynthia Lemere, Ph.D.
- David Knopman, M.D.
- Lee A. Jennings, M.D. MSHS
- Karen Bell, M.D.
- Lea Grinberg, M.D., Ph.D.
- Malú Tansey, Ph.D.
- Mary Sano, Ph.D.
- Oscar Lopez, M.D.
- Suzanne Craft, Ph.D.
- About Our Grants
- Andrew Kiselica, Ph.D., ABPP-CN
- Arjun Masurkar, M.D., Ph.D.
- Benjamin Combs, Ph.D.
- Charles DeCarli, M.D.
- Damian Holsinger, Ph.D.
- David Soleimani-Meigooni, Ph.D.
- Donna M. Wilcock, Ph.D.
- Elizabeth Head, M.A, Ph.D.
- Fan Fan, M.D.
- Fayron Epps, Ph.D., R.N.
- Ganesh Babulal, Ph.D., OTD
- Hui Zheng, Ph.D.
- Jason D. Flatt, Ph.D., MPH
- Jennifer Manly, Ph.D.
- Joanna Jankowsky, Ph.D.
- Luis Medina, Ph.D.
- Marcello D’Amelio, Ph.D.
- Marcia N. Gordon, Ph.D.
- Margaret Pericak-Vance, Ph.D.
- María Llorens-Martín, Ph.D.
- Nancy Hodgson, Ph.D.
- Shana D. Stites, Psy.D., M.A., M.S.
- Walter Swardfager, Ph.D.
- ALZ WW-FNFP Grant
- Capacity Building in International Dementia Research Program
- ISTAART IGPCC
- Alzheimer’s Disease Strategic Fund: Endolysosomal Activity in Alzheimer’s (E2A) Grant Program
- Imaging Research in Alzheimer’s and Other Neurodegenerative Diseases
- Zenith Fellow Awards
- National Academy of Neuropsychology & Alzheimer’s Association Funding Opportunity
- Part the Cloud-Gates Partnership Grant Program: Bioenergetics and Inflammation
- Pilot Awards for Global Brain Health Leaders (Invitation Only)
- Robert W. Katzman, M.D., Clinical Research Training Scholarship
- Funded Studies
- How to Apply
- Portfolio Summaries
- Supporting Research in Health Disparities, Policy and Ethics in Alzheimer’s Disease and Dementia Research (HPE-ADRD)
- Diagnostic Criteria & Guidelines
- Annual Conference: AAIC
- Professional Society: ISTAART
- Alzheimer's & Dementia
- Alzheimer's & Dementia: DADM
- Alzheimer's & Dementia: TRCI
- International Network to Study SARS-CoV-2 Impact on Behavior and Cognition
- Alzheimer’s Association Business Consortium (AABC)
- Global Biomarker Standardization Consortium (GBSC)
- Global Alzheimer’s Association Interactive Network
- International Alzheimer's Disease Research Portfolio
- Alzheimer’s Disease Neuroimaging Initiative Private Partner Scientific Board (ADNI-PPSB)
- Research Roundtable
- About WW-ADNI
- North American ADNI
- European ADNI
- Australia ADNI
- Taiwan ADNI
- Argentina ADNI
- WW-ADNI Meetings
- Submit Study
- RFI Inclusive Language Guide
- Scientific Conferences
- AUC for Amyloid and Tau PET Imaging
- Make a Donation
- Walk to End Alzheimer's
- The Longest Day
- RivALZ to End ALZ
- Ride to End ALZ
- Tribute Pages
- Gift Options to Meet Your Goals
- Founders Society
- Fred Bernhardt
- Anjanette Kichline
- Lori A. Jacobson
- Pam and Bill Russell
- Gina Adelman
- Franz and Christa Boetsch
- Adrienne Edelstein
- For Professional Advisors
- Free Planning Guides
- Contact the Planned Giving Staff
- Workplace Giving
- Do Good to End ALZ
- Donate a Vehicle
- Donate Stock
- Donate Cryptocurrency
- Donate Gold & Sterling Silver
- Donor-Advised Funds
- Use of Funds
- Giving Societies
- Why We Advocate
- Ambassador Program
- About the Alzheimer’s Impact Movement
- Research Funding
- Improving Care
- Support for People Living With Dementia
- Public Policy Victories
- Planned Giving
- Community Educator
- Community Representative
- Support Group Facilitator or Mentor
- Faith Outreach Representative
- Early Stage Social Engagement Leaders
- Data Entry Volunteer
- Tech Support Volunteer
- Other Local Opportunities
- Visit the Program Volunteer Community to Learn More
- Become a Corporate Partner
- A Family Affair
- A Message from Elizabeth
- The Belin Family
- The Eliashar Family
- The Fremont Family
- The Freund Family
- Jeff and Randi Gillman
- Harold Matzner
- The Mendelson Family
- Patty and Arthur Newman
- The Ozer Family
- Salon Series
- No Shave November
- Other Philanthropic Activities
- Still Alice
- The Judy Fund E-blast Archive
- The Judy Fund in the News
- The Judy Fund Newsletter Archives
- Sigma Kappa Foundation
- Alpha Delta Kappa
- Parrot Heads in Paradise
- Tau Kappa Epsilon (TKE)
- Sigma Alpha Mu
- Alois Society Member Levels and Benefits
- Alois Society Member Resources
- Zenith Society
- Founder's Society
- Joel Berman
- JR and Emily Paterakis
- Legal Industry Leadership Council
- Accounting Industry Leadership Council
Find Local Resources
Let us connect you to professionals and support options near you. Please select an option below:
Use Current Location Use Map Selector
Search Alzheimer’s Association
Our free ALZ Talks webinars provide education, information, news and resources on a variety of dementia and caregiving topics. Register for an ALZ Talks webinar — brought to you by the Alzheimer's Association, with support from Procter & Gamble.
ALZ Talks: Taking Charge of Your Brain Health
Building healthy habits into your everyday routines can lower the risk of cognitive decline and possibly dementia. Our guest speakers will share how they are prioritizing their brain health and give practical tips for keeping up healthy habits like physical activity, quality sleep, and controlling blood pressure. It's never too early or too late to take charge of your brain health! This webinar is supported by Procter & Gamble.

Missed a past ALZ Talks webinar?
View previous alz talks webinars on the alzheimer’s association youtube channel., the alzheimer’s association is in your community., locate dementia resources, programs and services in your area..
Find Community Resources
Connect with our free, online caregiver community.
Join ALZConnected
Keep Up With Alzheimer’s News and Events
- Patient Care & Health Information
- Diseases & Conditions
- Alzheimer's disease
An important part of diagnosing Alzheimer's disease includes being able to explain your symptoms. Input from a close family member or friend about your symptoms and their impact on your daily life helps. Tests of memory and thinking skills also help diagnose Alzheimer's disease.
Blood and imaging tests can rule out other potential causes of the symptoms. Or they may help your health care professional better identify the disease causing dementia symptoms.
In the past, Alzheimer's disease was diagnosed for certain only after death when looking at the brain with a microscope revealed plaques and tangles. Health care professional and researchers are now able to diagnose Alzheimer's disease during life with more certainty. Biomarkers can detect the presence of plaques and tangles. Biomarker tests include specific types of PET scans and tests that measure amyloid and tau proteins in the fluid part of blood and cerebral spinal fluid.
Diagnosing Alzheimer's disease would likely include the following tests:
Physical and neurological exam
A health care professional will perform a physical exam. A neurological exam may include testing:
- Muscle tone and strength.
- Ability to get up from a chair and walk across the room.
- Sense of sight and hearing.
- Coordination.
Blood tests may help rule out other potential causes of memory loss and confusion, such as a thyroid disorder or vitamin levels that are too low. Blood tests also can measure levels of beta-amyloid protein and tau protein, but these tests aren't widely available and coverage may be limited.
Mental status and neuropsychological testing
Your health care professional may give you a brief mental status test to assess memory and other thinking skills. Longer forms of this type of test may provide more details about mental function that can be compared with people of a similar age and education level. These tests can help establish a diagnosis and serve as a starting point to track symptoms in the future.
Brain imaging

- Brain scan images for diagnosis of Alzheimer's disease
Brain scans (FDG PET) used in the diagnosis of Alzheimer's disease. The scans show a healthy brain, a brain with mild cognitive impairment and a brain with Alzheimer's disease. Areas that are black and blue represent healthy brain metabolism. Areas that are green, yellow and red represent worsening brain metabolism as the disease progresses.
Images of the brain are typically used to pinpoint visible changes related to conditions other than Alzheimer's disease that may cause similar symptoms, such as strokes, trauma or tumors. New imaging techniques may help detect specific brain changes caused by Alzheimer's, but they're used mainly in major medical centers or in clinical trials.
Imaging of brain structures include:
- Magnetic resonance imaging (MRI). MRI uses radio waves and a strong magnetic field to produce detailed images of the brain. While they may show shrinkage of some brain regions associated with Alzheimer's disease, MRI scans also rule out other conditions. An MRI is generally preferred to a CT scan to evaluate dementia.
- Computerized tomography (CT). A CT scan, a specialized X-ray technology, produces cross-sectional images of your brain. It's usually used to rule out tumors, strokes and head injuries.
Positron emission tomography (PET) can capture images of the disease process. During a PET scan, a low-level radioactive tracer is injected into the blood to reveal a particular feature in the brain. PET imaging may include:
- Fluorodeoxyglucose (FDG) PET imaging scans show areas of the brain in which nutrients are poorly metabolized. Finding patterns in the areas of low metabolism can help distinguish between Alzheimer's disease and other types of dementia.
- Amyloid PET imaging can measure the burden of amyloid deposits in the brain. This test is mainly used in research but may be used if a person has unusual or very early onset of dementia symptoms.
- Tau PET imaging, which measures the tangles in the brain, is generally used in the research setting.
In special circumstances, other tests may be used to measure amyloid and tau in the cerebrospinal fluid. This may be done if symptoms are quickly getting worse or if dementia is affecting someone at a younger age than what's typical.
Future diagnostic tests
Researchers are working to develop tests that can measure biological signs of disease processes in the brain.
These tests, including blood tests, may improve accuracy when making a diagnosis. They also may allow the disease to be diagnosed before symptoms begin. A blood test to measure beta-amyloid levels is currently available.
Genetic testing isn't recommended for most people being evaluated for Alzheimer's disease. But people with a family history of early-onset Alzheimer's disease may consider it. Meet with a genetic counselor to discuss the risks and benefits before getting a genetic test.
- Care at Mayo Clinic
Our caring team of Mayo Clinic experts can help you with your Alzheimer's disease-related health concerns Start Here
More Information
Alzheimer's disease care at Mayo Clinic
- Diagnosing Alzheimer's
- Positron emission tomography scan
- Mayo Clinic Minute: New definition of Alzheimer's changes
Medications
Alzheimer's medicines can help with memory symptoms and other cognitive changes. Two types of drugs are currently used to treat symptoms:
Cholinesterase inhibitors. These medicines work by boosting levels of cell-to-cell communication. The medicines preserve a chemical messenger that is depleted in the brain by Alzheimer's disease. These are usually the first medicines tried, and most people see modest improvements in symptoms.
Cholinesterase inhibitors may improve symptoms related to behavior, such as agitation or depression. The medicines are taken orally or delivered through a patch on the skin. Commonly prescribed cholinesterase inhibitors include donepezil (Aricept, Adlarity), galantamine (Razadyne) and rivastigmine transdermal patch (Exelon).
The main side effects of these drugs include diarrhea, nausea, loss of appetite and sleep disturbances. In people with certain heart disorders, serious side effects may include an irregular heartbeat.
- Memantine (Namenda). This medicine works in another brain cell communication network and slows the progression of symptoms with moderate to severe Alzheimer's disease. It's sometimes used in combination with a cholinesterase inhibitor. Relatively rare side effects include dizziness and confusion.
In 2023, the U.S. Food and Drug Administration (FDA) approved lecanemab (Leqembi) for people with mild Alzheimer's disease and mild cognitive impairment due to Alzheimer's disease.
A phase 3 clinical trial found that the medicine slowed cognitive decline in people with early Alzheimer's disease. The medicine prevents amyloid plaques in the brain from clumping. The phase 3 trial was the largest so far to study whether clearing clumps of amyloid plaques from the brain can slow the disease.
Lecanemab is given as an IV infusion every two weeks. Side effects of lecanemab include infusion-related reactions such as fever, flu-like symptoms, nausea, vomiting, dizziness, changes in heart rate and shortness of breath.
Also, people taking lecanemab may have swelling in the brain or may get small bleeds in the brain. Rarely, brain swelling can be serious enough to cause seizures and other symptoms. Also in rare instances, bleeding in the brain can cause death. The FDA recommends getting a brain MRIs before starting treatment. It also recommends being monitored with brain MRI during treatment for symptoms of brain swelling or bleeding.
People who carry a certain form of a gene known as APOE e4 appear to have a higher risk of these serious complications. The FDA recommends being tested for this gene before starting treatment with lecanemab.
If you take a blood thinner or have other risk factors for brain bleeding, talk to your health care professional before taking lecanemab. Blood-thinning medicines may increase the risk of bleeds in the brain.
More research is being done on the potential risks of taking lecanemab. Other research is looking at how effective lecanemab may be for people at risk of Alzheimer's disease, including people who have a first-degree relative, such as a parent or sibling, with the disease.
Another medicine being studied is donanemab. It targets and reduces amyloid plaques and tau proteins. It was found to slow declines in thinking and functioning in people with early Alzheimer's disease.
Sometimes other medicines such as antidepressants may be prescribed to help control the behavioral symptoms associated with Alzheimer's disease.
Creating a safe and supportive environment
An important part of any treatment plan is to adapt to the needs of a person with Alzheimer's disease. Establish and strengthen routine habits and cut down on tasks that require memory. These steps can make life much easier.
These are ways to support a person's sense of well-being and continued ability to function:
- Keep keys, wallets, mobile phones and other valuables in the same place at home so they don't become lost.
- Keep medicines in a secure location. Use a daily checklist to keep track of doses.
- Arrange for finances to be on automatic payment and automatic deposit.
- Have the person with Alzheimer's carry a mobile phone with location tracking. Program important phone numbers into the phone.
- Install alarm sensors on doors and windows.
- Make sure regular appointments are on the same day at the same time as much as possible.
- Use a calendar or whiteboard to track daily schedules. Build the habit of checking off completed items.
- Remove excess furniture, clutter and throw rugs.
- Install sturdy handrails on stairs and in bathrooms.
- Ensure that shoes and slippers are comfortable and provide good traction.
- Reduce the number of mirrors. People with Alzheimer's may find images in mirrors confusing or scary.
- Make sure that the person with Alzheimer's carries ID or wears a medical alert bracelet.
- Keep photos and other objects with meaning around the house.
- Alzheimer's drugs
- Alzheimer's: New treatments
- Lecanemab for Alzheimer's disease
- Video: Alzheimer's drug shows early promise
Clinical trials
Explore Mayo Clinic studies testing new treatments, interventions and tests as a means to prevent, detect, treat or manage this condition.
Alternative medicine
Herbal remedies, vitamins and other supplements are widely promoted for cognitive health or to prevent or delay Alzheimer's. But clinical trials have produced mixed results. There's little evidence to support them as effective treatments.
Some of the treatments that have been studied recently include:
Vitamin E. Although vitamin E doesn't prevent Alzheimer's, taking 2,000 international units daily may help delay symptoms getting worse in people who already have mild to moderate disease. However, study results have been mixed, with only some showing modest benefits. Further research into the safety of 2,000 international units daily of vitamin E in a dementia population will be needed before it can be routinely recommended.
Supplements promoted for cognitive health can interact with medicines you're taking for Alzheimer's disease or other health conditions. Work closely with your health care team to create a safe treatment plan. Tell your health care team about your prescriptions and any medicines or supplements you take without a prescription.
- Omega-3 fatty acids. Omega-3 fatty acids in fish or from supplements may lower the risk of developing dementia. But clinical studies have shown no benefit for treating Alzheimer's disease symptoms.
- Curcumin. This herb comes from turmeric and has anti-inflammatory and antioxidant properties that might affect chemical processes in the brain. So far, clinical trials have found no benefit for treating Alzheimer's disease.
- Ginkgo. Ginkgo is a plant extract. A large study funded by the National Institutes of Health found no effect in preventing or delaying Alzheimer's disease.
- Melatonin. This supplement helps regulate sleep. It's being studied to see if it can help people with dementia manage sleep problems. But some research has indicated that melatonin may worsen mood in some people with dementia. More research is needed.
Lifestyle and home remedies
Healthy lifestyle choices promote good overall health. They also may play a role in maintaining brain health.
Regular exercise is an important part of a treatment plan. Activities such as a daily walk can help improve mood and maintain the health of joints, muscles and the heart. Exercise also promotes restful sleep and prevents constipation. It's beneficial for care partners, too.
People with Alzheimer's who have trouble walking may still be able to use a stationary bike, stretch with elastic bands or participate in chair exercises. You may find exercise programs geared to older adults on TV, the internet or DVDs.
People with Alzheimer's may forget to eat, lose interest in meals or may not eat healthy foods. They may also forget to drink enough, leading to dehydration and constipation.
Offer the following:
- Healthy options. Buy favorite healthy food options that are easy to eat.
- Water and other healthy beverages. Encourage drinking several glasses of liquids every day. Avoid beverages with caffeine, which can increase restlessness, interfere with sleep and trigger a need to urinate often.
- High-calorie, healthy shakes and smoothies. Serve milkshakes with protein powders or make smoothies. This is helpful when eating becomes more difficult.
Social engagement and activities
Social activities can support preserved skills and abilities. They also help with over-all well-being. Do things that are meaningful and enjoyable. Someone with dementia might:
- Listen to music or dance.
- Read or listen to books.
- Garden or do crafts.
- Go to social events at senior or memory care centers.
- Do activities with children.
Coping and support
People with Alzheimer's disease experience a mixture of emotions — confusion, frustration, anger, fear, uncertainty, grief and depression.
If you're caring for someone with Alzheimer's, you can help them cope by being there to listen. Reassure the person that life can still be enjoyed, provide support, and do your best to help the person retain dignity and self-respect.
A calm and stable home environment can help reduce behavior problems. New situations, noise, large groups of people, being rushed or pressed to remember, or being asked to do complex tasks can cause anxiety. As a person with Alzheimer's becomes upset, the ability to think clearly declines even more.
Caring for the caregiver
Caring for a person with Alzheimer's disease is physically and emotionally demanding. Feelings of anger, guilt, stress, worry, grief and social isolation are common.
Caregiving can even take a toll on the caregiver's physical health. Pay attention to your own needs and well-being. It's one of the most important things you can do for yourself and for the person with Alzheimer's.
If you're a caregiver for someone with Alzheimer's, you can:
- Learn as much about the disease as you can.
- Ask questions of health care professionals, social workers and others involved in the care of your loved one.
- Call on friends or other family members for help when you need it.
- Take a break every day.
- Spend time with your friends.
- Take care of your health by seeing your own health care professionals on schedule, eating healthy meals and getting exercise.
- Join a support group.
- Make use of a local adult day center, if possible.
Many people with Alzheimer's and their families benefit from counseling or local support services. Contact your local Alzheimer's Association affiliate to connect with support groups, health care professionals, occupational therapists, resources and referrals, home care agencies, residential care facilities, a telephone help line, and educational seminars.
Preparing for your appointment
Medical care for the loss of memory or other thinking skills usually requires a team or partner strategy. If you're worried about memory loss or related symptoms, ask a close relative or friend to go with you to an appointment with a health care professional. In addition to providing support, your partner can provide help in answering questions.
If you're going with someone to a health care appointment, your role may be to provide some history or your thoughts on changes you have seen. This teamwork is an important part of medical care.
Your health care professional may refer you to a neurologist, psychiatrist, neuropsychologist or other specialist for further evaluation.
What you can do
You can prepare for your appointment by writing down as much information as possible to share. Information may include:
- Medical history, including any past or current diagnoses and family medical history.
- Medical team, including the name and contact information of any current physician, mental health professional or therapist.
- Medicines, including prescriptions, medicines you take without a prescription, vitamins, herbs or other supplements.
- Symptoms, including specific examples of changes in memory or thinking skills.
What to expect from your doctor
Your health care professional will likely ask a number of questions to understand changes in memory or other thinking skills. If you are accompanying someone to an appointment, be prepared to provide your thoughts as needed. Your care professional may ask:
- What kinds of memory trouble and mental lapses are you having? When did you first notice them?
- Are they steadily getting worse, or are they sometimes better and sometimes worse?
- Have you stopped doing certain activities, such as managing finances or shopping, because these activities were too mentally challenging?
- How is your mood? Do you feel depressed, sadder or more anxious than usual?
- Have you gotten lost lately while driving or in a situation that's usually familiar to you?
- Has anyone expressed unusual concern about your driving?
- Have you noticed any changes in the way you tend to react to people or events?
- Do you have more energy than usual, less than usual or about the same?
- What medicines are you taking? Are you taking any vitamins or supplements?
- Do you drink alcohol? How much?
- Have you noticed any trembling or trouble walking?
- Are you having trouble remembering health care appointments or when to take your medicines?
- Have you had your hearing and vision tested recently?
- Did anyone else in your family ever have memory trouble? Was anyone ever diagnosed with Alzheimer's disease or dementia?
- Do you act out your dreams while sleeping (punch, flail, shout, scream)? Do you snore?
- Jankovic J, et al., eds. Alzheimer disease and other dementias. In: Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Elsevier; 2022. https://www.clinicalkey.com. Accessed Sept. 21, 2022.
- Alzheimer's disease fact sheet. National Institute on Aging. https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet. Accessed Oct. 3, 2022.
- Livingston G, et al. Dementia prevention, intervention, and care: 2020 report of The Lancet Commission. The Lancet. 2020; doi:10.1016/S0140-6736(20)30367-6.
- Chen L, et al. Plasma tau proteins for the diagnosis of mild cognitive impairment and Alzheimer's disease: A systematic review and meta-analysis. Frontiers in Aging Neuroscience. 2022; doi:10.3389/fnagi.2022.942629.
- Dementia. World Health Organization. https://www.who.int/news-room/fact-sheets/detail/dementia. Accessed Oct. 3, 2022.
- Alzheimer's disease and related dementias. Centers for Disease Control and Prevention. https://www.cdc.gov/aging/aginginfo/alzheimers.htm. Accessed Oct. 3, 2022.
- What is dementia? Symptoms, types, and diagnosis. National Institute on Aging. https://www.nia.nih.gov/health/diagnosing-dementia. Accessed Oct. 3, 2022.
- Biomarkers for dementia detection and research. National Institute on Aging. https://www.nia.nih.gov/health/biomarkers-dementia-detection-and-research. Accessed Oct. 3, 2022.
- Middle-stage caregiving. Alzheimer's Association. https://www.alz.org/help-support/caregiving/stages-behaviors/middle-stage. Accessed Oct. 3, 2022.
- Kellerman RD, et al. Alzheimer's disease. In: Conn's Current Therapy 2022. Elsevier; 2022. https://www.clinicalkey.com. Accessed Sept. 21, 2022.
- Ferri FF. Alzheimer disease. In: Ferri's Clinical Advisor 2023. Elsevier; 2023. https://www.clinicalkey.com. Accessed Oct. 3, 2022.
- Early-stage caregiving. Alzheimer's Association. https://www.alz.org/help-support/caregiving/stages-behaviors/early-stage. Accessed Oct. 3, 2022.
- Alzheimer's disease at a glance. National Center for Complementary and Integrative Health. https://nccih.nih.gov/health/alzheimer/ataglance. Accessed Oct. 3, 2022.
- Budson AE, et al. Alzheimer's disease. In: Memory Loss, Alzheimer's Disease, and Dementia. 3rd ed. Elsevier; 2022. https://www.clinicalkey.com. Accessed Oct. 3, 2022.
- Coping with late-stage Alzheimer's disease. National Institute on Aging. https://www.nia.nih.gov/health/coping-late-stage-alzheimers-disease. Accessed Oct. 3, 2022.
- Levenson JL, ed. Dementia. In: The American Psychiatric Association Publishing Textbook of Psychosomatic Medicine and Consultation-Liaison Psychiatry. 3rd ed. American Psychiatric Association Publishing; 2019. https://psychiatryonline.org. Accessed Oct. 3, 2022.
- Dementia: Lifestyle and management. American Geriatrics Society. https://www.healthinaging.org/a-z-topic/dementia/lifestyle. Accessed Oct. 5, 2022.
- Is Alzheimer's genetic? Alzheimer's Association. https://www.alz.org/alzheimers-dementia/what-is-alzheimers/causes-and-risk-factors/genetics. Accessed Oct. 6, 2022.
- Ami T. Allscripts EPSi. Mayo Clinic. July 6, 2022.
- Alzheimer's disease research centers. National Institute on Aging. https://www.nia.nih.gov/health/alzheimers-disease-research-centers#minnesota. Accessed Sept. 26, 2022.
- About the Alzheimer's Consortium. Arizona Alzheimer's Consortium. https://azalz.org/about/#institutes. Accessed Sept. 26, 2022.
- Graff-Radford J (expert opinion). Mayo Clinic. Oct. 6, 2022.
- Shi M, et al. Impact of anti-amyloid-β monoclonal antibodies on the pathology and clinical profile of Alzheimer's disease: A focus on aducanumab and lecanemab. Frontiers in Aging and Neuroscience. 2022; doi:10.3389/fnagi.2022.870517.
- Cummings J, et al. Alzheimer's disease drug development pipeline: 2022. Alzheimer's and Dementia. 2022; doi:10.1002/trc2.12295.
- Leqembi (prescribing information). Eisai Inc.; 2023. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=761269. Accessed July 10, 2023.
- Mintun MA, et al. Donanemab in early Alzheimer's disease. New England Journal of Medicine. 2021; doi:10.1056/NEJMoa2100708.
- 2022 Alzheimer's disease facts and figures. Alzheimer's Association. https://www.alz.org/alzheimers-dementia/facts-figures. Accessed Oct. 4, 2022.
- Rosenberg A, et al. Multidomain interventions to prevent cognitive impairment, Alzheimer's disease and dementia: From FINGER to world-wide FINGERS. The Journal of Prevention of Alzheimer's Disease. 2020; doi:10.14283/jpad.2019.41.
- Bleicher K, et al. Cohort profile update: The 45 and up study. International Journal of Epidemiology. 2022; doi:10.1093/ije/dyac104.
- Leqembi (approval letter). Biologic License Application 761269. U.S. Food and Drug Administration. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=761269. Accessed July 7, 2023.
- Swaddiwudhipong N, et al. Pre-diagnostic cognitive and functional impairment in multiple sporadic neurodegenerative diseases. Alzheimer's and Dementia. 2022; doi:10.1002/alz.12802.
- Van Dyck CH, et al. Lecanemab in early Alzheimer's disease. New England Journal of Medicine. 2023; doi:10.1056/NEJMoa2212948.
- Alzheimer's 101
- Alzheimer's genes
- Alzheimer's prevention: Does it exist?
- Alzheimer's stages
- Brain atrophy and Alzheimer's
- Did the definition of Alzheimer's disease change?
- Healthy brain and brain with Alzheimer's disease
- Mayo Clinic Minute: 3 tips to reduce your risk of Alzheimer's disease
- Mayo Clinic Minute: Alzheimer's disease risk and lifestyle
- Mayo Clinic Minute: Women and Alzheimer's Disease
- Memory loss: When to seek help
- New Alzheimers Research
- Sundowning: Late-day confusion
- Understanding the difference between dementia types
- Young-onset Alzheimer's
Associated Procedures
News from mayo clinic.
- Mayo Clinic Minute: Reducing risk of Alzheimer's disease in families Nov. 13, 2023, 04:02 p.m. CDT
- Alzheimer's drug lecanemab granted full approval by FDA, Mayo Clinic expert weighs in July 06, 2023, 09:55 p.m. CDT
- Mayo Clinic expert talks benefits, risks of experimental Alzheimer's drug May 04, 2023, 04:17 p.m. CDT
- $41 million federal grant to help Mayo Clinic, collaborators advance multiethnic Alzheimer's research March 28, 2023, 03:00 p.m. CDT
- What is clinically meaningful to slow Alzheimer's progression? Feb. 07, 2023, 03:10 p.m. CDT
- Mayo Clinic expert provides perspective on FDA approval of another experimental Alzheimer's drug Jan. 06, 2023, 08:50 p.m. CDT
- Mayo Clinic Minute: Family risk of Alzheimer's disease Nov. 14, 2022, 04:25 p.m. CDT
- Mayo Clinic expert offers perspective on experimental Alzheimer's disease drug Sept. 29, 2022, 05:15 p.m. CDT
- Science Saturday: In neurodegenerative diseases, Mayo Clinic research finds shared gene patterns, widespread damage May 28, 2022, 11:00 a.m. CDT
Products & Services
- A Book: Day to Day: Living With Dementia
- A Book: Mayo Clinic on Alzheimer's Disease
- A Book: Mayo Clinic on Healthy Aging
- Assortment of Products for Independent Living from Mayo Clinic Store
- Symptoms & causes
- Diagnosis & treatment
- Doctors & departments
Mayo Clinic does not endorse companies or products. Advertising revenue supports our not-for-profit mission.
- Opportunities
Mayo Clinic Press
Check out these best-sellers and special offers on books and newsletters from Mayo Clinic Press .
- Mayo Clinic on Incontinence - Mayo Clinic Press Mayo Clinic on Incontinence
- The Essential Diabetes Book - Mayo Clinic Press The Essential Diabetes Book
- Mayo Clinic on Hearing and Balance - Mayo Clinic Press Mayo Clinic on Hearing and Balance
- FREE Mayo Clinic Diet Assessment - Mayo Clinic Press FREE Mayo Clinic Diet Assessment
- Mayo Clinic Health Letter - FREE book - Mayo Clinic Press Mayo Clinic Health Letter - FREE book
Make twice the impact
Your gift can go twice as far to advance cancer research and care!
Ohio State nav bar
The Ohio State University
- BuckeyeLink
- Find People
- Search Ohio State
PATHOPHYSIOLOGY AND CLINICAL PRESENTATION – CORRECT DIAGNOSIS
Alzheimer’s disease (AD) is characterized by two pathological lesions, beta-amyloid (Aβ) plaques and neurofibrillary tangles, that are progressively distributed throughout the brain. These lesions are associated with disruption of the communication pathways between neurons, neuron degeneration, brain atrophy, and functional loss. However, the specific etiology of AD remains uncertain (Sperling et al, 2011).
Photomicrograph of human brain with Alzheimer disease following Beilchowsky stain. The stain highlights an extracellular neuritic plaque (upper right) and an intraneuronal neurofibrillary tangle (lower left) (Up to Date).

With synaptic activation: (1) intraneuronal levels of Aβ (red) are reduced, (2) Aβ secretion is augmented, (3) neprilysin-induced Aβ degradation is enhanced, (4) transcription of pro-survival genes (AID) increases, (5) local ATP synthesis rises at synapses, and (6) tau secretion is augmented (Tampellini, 2015).
Beta-amyloid plaques
The outer cell membrane of neurons contains an amyloid precursor protein (APP). In the normal brain, alpha-secretase and gamma-secretase cut APP into smaller protein fragments that are thought to be helpful to neurons and then later cleared from the brain. In AD, beta-secretase and gamma-secretase cut APP into small Aβ fragments. Researchers hypothesize that the Aβ oligomers disrupt neuronal cell signaling and communication at the synapses and later stick together to form beta-amyloid plaques (NIA, 2015). Researchers believe the beta-amyloid plaques may also trigger an inflammatory immune reaction that results in neuronal death. In early onset familiar AD, the formation of Aβ plaques is related to autosomal dominant mutations of the APP gene, presenilin 1, and presenilin 2 genes. Apolipoprotein E gene-allel4 may play a role in Aβ plaque formation in late onset AD (McCance & Huether, 2014).
Neurofibrilary tangles
Neurons have microtubules that transport nutrients and other substances. In the normal brain, tau proteins provide stability and keep the microtubules straight. In AD, tau proteins are hyperphosphorylated and disassociate from the microtubules. The tau proteins accumulate inside the neuron, forming neurofibrillary tangles that impede nutrient transport and disrupt communication (NIA, 2015). These tangles are toxic to the neuron and eventually contribute to neuronal death.
Video from http://ed.ted.com/lessons/what-is-alzheimer-s-disease-ivan-seah-yu-jun
Progressive Spread of Lesions and Stages of Disease
While both types of lesions result in cell death and brain atrophy, neurofibrillary tangles appear to spread throughout the brain in an orderly procession that corresponds to the stages of Alzheimers disease progression (Keene, et al, 2015). Neurofibrilary tangles begin in the hippocampus and then spread throughout the brain before finally developing in the medullary centers leading to death. (Granic, et. Al, 2010). Lesions can develop in the brain 10-20 years before symptoms appear and progression of the lesions throughout the brain can occur for as long as 10 years (NIA, 2015).
Video from http://aboutalz.org/%23
Leave a reply cancel reply.
Your email address will not be published. Required fields are marked *
Save my name, email, and website in this browser for the next time I comment.
Innovative microscopy demystifies metabolism of Alzheimer's
Alzheimer's disease causes significant problems with memory, thinking and behavior and is the most common form of dementia, affecting more than 50 million people around the world each year. This number is expected to triple by the year 2050.
Using their own state-of-the art imaging technologies, scientists at the University of California San Diego have now revealed how the metabolism of lipids, a class of molecule that includes fats, oils and many hormones, is changed in Alzheimer's disease. They also revealed a new strategy to target this metabolic system with new and existing drugs. The findings are published in Cell Metabolism .
"Lipids have been associated with Alzheimer's for as long as we've known about the disease," said senior and co-corresponding author Xu Chen, Ph.D., an assistant professor in the Department of Neurosciences at UC San Diego School of Medicine, referring to the original 1907 report by Alois Alzheimer that described the unusual presence of fat deposits in the brain of the first person to be diagnosed with the disease. "So much of the emphasis since then has been placed on tau and other proteins that the research community has, until the last decade or so, largely overlooked this important aspect of the disease."
"Driven by a keen interest in lipid droplet functions in aging and disease, we initiated this fruitful collaboration to harness cutting-edge SRS technology for studying lipid metabolism in tauopathy mouse brains." Said Yajuan Li, M.D., Ph.D., a postdoctoral researcher in the Shu Chien-Gene Lay Department of Bioengineering at UC San Diego Jacobs School of Engineering. SRS imaging is an approach that analyzes the way molecules in a sample interact with laser light.
In the brain, lipids come in the form of tiny droplets that control a variety of processes, such as energy storage and cellular responses to stress. These processes are tightly regulated in typical brains, but in Alzheimer's or similar diseases, lipid droplet metabolism can malfunction. While scientists understand that there is a relationship between Alzheimer's and lipid metabolism, exactly how they influence one another has remained a mystery.
To answer this question, the team looked directly at lipid droplets in the brains of mice with excess tau protein. They used a state-of-the-art SRS imaging platform developed in Lingyan Shi's lab at the Jacobs School of Engineering. The platform makes it possible to take microscopic images of lipid droplets within cells without the use chemical dyes, which can alter the delicate molecules and compromise the results.
"Intriguingly, the inert lipid droplets observed in tauopathy brains exhibit similar behavior to those found in aging brains," said co-corresponding author Lingyan Shi, Ph.D., assistant professor of bioengineering at the Jacobs School. "We are now focusing on understanding the underlying mechanisms by combing SRS imaging with other utilizing multidisciplinary techniques. Our approach is biologically neutral, so we're able to observe what's happening in the brain at the molecular level with as little interference as possible."
Shi and her team, including Li, pioneered the new approach, which uses a specially modified version of water, called heavy water, as a metabolic probe.
"Instead of using a typical chemical dye to stain lipids, we use heavy water that is naturally participating in the metabolic activities we're interested in," added Shi. "This gives us a much clearer picture of how lipids are formed spatiotemporally, which would not be possible with other approaches. Our current focus is on comprehending the underlying mechanisms of these dynamic changes of lipid metabolism in the context of aging and diseases."
The researchers discovered that in brains with tauopathy, neurons accumulate excess lipids as a result of stress or damage. This influx forces neurons to offload the excess to immune cells in the brain, called microglia. These microglia then mount an inflammatory response that causes further stress to neurons, triggering a repeating and worsening cycle.
In addition to characterizing this process, they were also able to identify a critical enzyme, called adenosine monophosphate-activated protein kinase (AMPK) that orchestrates the cycle. According to the researchers, breaking this cycle could unlock new treatment options for Alzheimer's disease. Chen is particularly optimistic about the possibility of repurposing existing drugs that modify lipid metabolism.
"We don't think this is an incidental phenomenon," said Chen. "The evidence suggests that lipid metabolism is a driving mechanism for Alzheimer's disease. There are many drugs that target lipid metabolism in other body systems, such as in the liver, so we might be able to change this system quite dramatically using tools we already have."
- Alzheimer's Research
- Healthy Aging
- Gene Therapy
- Diseases and Conditions
- Alzheimer's
- Disorders and Syndromes
- Alzheimer's disease
- Dementia with Lewy bodies
- Confocal laser scanning microscopy
- Transmission electron microscopy
- Psychotherapy
- Urinary incontinence
- Erectile dysfunction
- Biological tissue
Story Source:
Materials provided by University of California - San Diego . Original written by Miles Martin. Note: Content may be edited for style and length.
Journal Reference :
- Yajuan Li, Daniel Munoz-Mayorga, Yuhang Nie, Ningxin Kang, Yuren Tao, Jessica Lagerwall, Carla Pernaci, Genevieve Curtin, Nicole G. Coufal, Jerome Mertens, Lingyan Shi, Xu Chen. Microglial lipid droplet accumulation in tauopathy brain is regulated by neuronal AMPK . Cell Metabolism , 2024; DOI: 10.1016/j.cmet.2024.03.014
Cite This Page :
Explore More
- Humans and Earth's Deep Subsurface Fluid Flow
- Holographic Displays: An Immersive Future
- Harvesting Energy Where River Meets Sea
- Making Diamonds at Ambient Pressure
- Eruption of Mega-Magnetic Star
- Clean Fuel Generation With Simple Twist
- Bioluminescence in Animals 540 Million Years Ago
- Fossil Frogs Share Their Skincare Secrets
- Fussy Eater? Most Parents Play Short Order Cook
- Precise Time Measurement: Superradiant Atoms
Trending Topics
Strange & offbeat.
April 23, 2024
“Partnership for the Life Course of Research”: Promoting Diversity in Alzheimer’s Research

Hispanic/Latino individuals are 1.5 times more likely to develop Alzheimer’s disease compared to their non-Hispanic white counterparts . Despite these disparities, studies consistently show that minoritized groups are underrepresented in research studies. This under-recruitment can not only perpetuate health disparities but also impact the generalizability of findings and effectiveness of Alzheimer’s disease treatments. To address this issue, the University of Washington’s Alzheimer’s Disease Research Center (ADRC) partnered with researchers from the Health Promotion Research Center (HPRC) to increase participation of all underrepresented groups in their ongoing Clinical Core research, starting with Latinx communities with whom the team had long-standing partnerships.
Principal investigator, Linda Ko , along with research scientist Sonia Bishop, research coordinators Katherine Lopéz and Janet Rojina, and graduate research assistant Kelley Pascoe of HPRC, came together to expand and grow the initial ADRC partnership with Hispanic and Latino communities in the Seattle area by continuing to build trust in the ADRC. “A lot of communities are really wary of joining research – and for good reason,” said Lopéz, referring to historical injustices and systemic discrimination within the world of research. “Having a connection to the community and making connections with people they already trust is necessary, especially when you’re working with older adults.”
A Community-Based Approach
Recognizing the limited capacity for language and culturally concordant team members, they made sure to build an intentionally bi-lingual and bi-cultural team. “It’s very important that our team look like the community we collaborate with, to create something by the community and for the community,” said Bishop. They also established a new Community Advisory Board (CAB) composed of trusted leaders from community-based organizations (CBOs). The CAB could vouch for the team’s intentions with community members and provide guidance and direction.

Together, the team and CAB launched a multi-pronged approach to build trust and garner interest and participation in the research project. “You have to do a little bit of everything to make sure you have a presence in the community,” said Rojina. “You want to make sure the community knows who we are when they see us and that we’re someone they trust.” The team attended local tabling events and partnered with CBOs to create new events for community members. This included Cafecito y Carla (Coffee Chats), a small, relaxed group setting where people could learn more about the research, ask questions, and get to know the team members. “The most important part of research is building relationships,” Rojina noted. “Once you have a relationship with the participants, they’ll be more willing to listen and better able to decide what’s best for themselves.”
The largest community event the team hosted was the Brain Health and Community Resource Fair, adapted from a previous event created by the National Alzheimer’s Coordinating Center (NACC) to increase Native American participants in AD research . The interactive event kicked off with an introductory presentation on Alzheimer’s disease and brain health from Dr. Karen Torres, a bi-cultural, bi-lingual neuropsychologist from the ADRC. Afterwards, attendees cycled through interactive learning stations where they could learn about brain health, nutrition, and physical activity. Participants could also engage in cognitive testing, get their blood pressure taken, and watch educational videos on AD. Additionally, the event had a station where participants could meet researchers, ask questions, and enroll in the research study, should they choose to.

Making Improvements
By leveraging events and building relationships and their presence in the community, the team created welcoming spaces that resonated with Hispanic/Latino communities. Over the course of a year, Hispanic/Latino enrollment in the research study increased from 3 individuals in 2022 to 26 in 2023. “Engaging underrepresented groups is not necessarily hard, it’s just different,” explained Rojina, “It takes time, patience, and flexibility; you have to be adaptable to meet the needs of the community.”
While the increase in participants was the desired outcome, Bishop explained that the real measurement of success was the trust built within the community. “We had a few people who were already enrolled in the study and had gone through their first visit, and they ended up coming to a Coffee Chat and bringing other family members who they thought could benefit from participating in the study. They understood the importance of the study and trusted us enough to invite their loved ones to be a part of this program. That’s really how research grows, through trust and word of mouth.”
Community interest in the project has only increased over time, and the team has found that there are more people who want to participate than they can currently accommodate. “We’re doing our best to meet the needs of the community in every way we can and with every ask that we get,” Bishop affirmed. “Even now at max capacity, we are transparent with where we’re at and tell community members that it could take a year or longer to get them into the study.” In the meantime, the team will continue their educational efforts and plans to host another Brain Health and Community Resource Fair in the Yakima area.
Looking towards the future, the team hopes to use the lessons and approaches learned during this project with other communities to continue expanding Alzheimer’s disease research. The CAB are vocal advocates for providing these research opportunities to all communities and hope to pivot to focus on Asian American and Pacific Islander communities next. Nationally, the languages for cognitive testing used in Alzheimer’s disease research are only in English and Spanish, which can make it difficult to include people who speak other languages in studies. While they advocate and wait for more translated testing materials, the team plans to continue educational events and to build trust.
Reflecting on their project, its successes, and the new challenges the team will face, Pascoe emphasized that future successes will come through time and with partnership. “Partnership is for the life course of research. It’s not just for recruitment or just having someone walk in the door and join the study. It’s partnering with them all the way to results, sharing results, making sure that those results make sense, and then asking them what other questions they have and what other directions do they want to take ,” she articulated. This comprehensive approach underscores the team’s commitment to community engagement beyond mere enrollment numbers.
Alzheimer’s is projected to affect 13.9 million Americans by 2060 . Diversity in research will be essential to ensure that findings and treatments are relevant to everyone. Through intentional relationship-building and inclusive practices, researchers can not only increase enrollment and make research more accessible, but foster trust and collaboration to create community-led solutions that are impactful for all.
© 2024 University of Washington | Seattle, WA
Got any suggestions?
We want to hear from you! Send us a message and help improve Slidesgo
Top searches
Trending searches

suicide prevention
8 templates

46 templates

tropical rainforest
29 templates

spring season
34 templates

american football
16 templates

32 templates
World Alzheimer's Day
World alzheimer's day presentation, free google slides theme and powerpoint template.
There's still time to download this template and create a presentation in honor of World Alzheimer's Day, which is celebrated on September 21. Since it's a disease that has no cure yet, we have used a design that eases the atmosphere and softens the mood. For example, we've included rounded shapes and very light tones of purple. There are also slides for informative purposes, as they contain tables or graphs where you can add data about this disease.
Features of this template
- 100% editable and easy to modify
- 35 different slides to impress your audience
- Contains easy-to-edit graphics such as graphs, maps, tables, timelines and mockups
- Includes 500+ icons and Flaticon’s extension for customizing your slides
- Uses illustrated concepts from Storyset : editable color, different backgrounds, animated illustrations
- Designed to be used in Google Slides and Microsoft PowerPoint
- 16:9 widescreen format suitable for all types of screens
- Includes information about fonts, colors, and credits of the free resources used
How can I use the template?
Am I free to use the templates?
How to attribute?
Attribution required If you are a free user, you must attribute Slidesgo by keeping the slide where the credits appear. How to attribute?
Related posts on our blog.

How to Add, Duplicate, Move, Delete or Hide Slides in Google Slides

How to Change Layouts in PowerPoint

How to Change the Slide Size in Google Slides
Related presentations.

Premium template
Unlock this template and gain unlimited access

- Today's news
- Reviews and deals
- Climate change
- 2024 election
- Fall allergies
- Health news
- Mental health
- Sexual health
- Family health
- So mini ways
- Unapologetically
- Buying guides
Entertainment
- How to Watch
- My Portfolio
- Stock Market
- Biden Economy
- EV Deep Dive
- Stocks: Most Actives
- Stocks: Gainers
- Stocks: Losers
- Trending Tickers
- World Indices
- US Treasury Bonds
- Top Mutual Funds
- Highest Open Interest
- Highest Implied Volatility
- Stock Comparison
- Advanced Charts
- Currency Converter
- Investment Ideas
- Research Reports
- Basic Materials
- Communication Services
- Consumer Cyclical
- Consumer Defensive
- Financial Services
- Industrials
- Real Estate
- Mutual Funds
- Credit Cards
- Balance transfer cards
- Cash-back cards
- Rewards cards
- Travel cards
- Personal Loans
- Student Loans
- Car Insurance
- Morning Brief
- Market Domination
- Market Domination Overtime
- Opening Bid
- Stocks in Translation
- Lead This Way
- Good Buy or Goodbye?
- Fantasy football
- Pro Pick 'Em
- College Pick 'Em
- Fantasy baseball
- Fantasy hockey
- Fantasy basketball
- Download the app
- Daily fantasy
- Scores and schedules
- GameChannel
- World Baseball Classic
- Premier League
- CONCACAF League
- Champions League
- Motorsports
- Horse racing
- Newsletters
New on Yahoo
- Privacy Dashboard
Yahoo Finance
Nkgen biotech announces upcoming presentation on snk01 nk cell therapy in neurodegenerative disease at the 12th annual alzheimer’s & parkinson’s drug development summit.
SANTA ANA, Calif., April 18, 2024 (GLOBE NEWSWIRE) -- NKGen Biotech, Inc. (Nasdaq: NKGN) (“NKGen” or the “Company”), a clinical-stage biotechnology company focused on the development and commercialization of innovative autologous, allogeneic and CAR-NK natural killer (“NK”) cell therapeutics, today announced that Paul Y. Song, MD, Chairman and Chief Executive Officer of NKGen Biotech, will present details around its NK cell therapy in neurodegenerative disease, review its preclinical data in Parkinson’s disease, and highlight clinical safety and efficacy data from the Company’s recently completed Phase 1 clinical trial of SNK01, autologous natural killer cell therapy, in Alzheimer’s disease at the 12 th Annual Alzheimer’s & Parkinson’s Drug Development Summit to be held in Boston, MA, from April 23–25, 2024.
Dr. Song’s presentation will detail how enhanced natural killer cells can maximize the body’s ability to destroy diseased and abnormal cells and provide a review of NK cell therapy as an alternative treatment modality to resolve neurodegeneration by harnessing the body’s natural immune system, including a discussion on the scientific rationale behind the use of NK cell therapy for the treatment of Parkinson’s disease. Dr. Song will also present clinical safety and efficacy data from the Company’s recently completed Phase 1 clinical trial evaluating autologous SNK01 NK cell therapy in patients with Alzheimer’s disease (NCT04678453).
A copy of the presentation will be available on the Scientific Publications page of the Company’s website at https://nkgenbiotech.com/ once the presentation has concluded. Previously disclosed Phase 1 data on the positive effects of SNK01 on amyloid, tau and neuroinflammation biomarkers, which may not be included in this conference presentation, can also be found on the Scientific Publications page.
About NKGen Biotech
NKGen is a clinical-stage biotechnology company focused on the development and commercialization of innovative autologous, allogeneic, and CAR-NK NK cell therapeutics. NKGen is headquartered in Santa Ana, California, USA. For more information, please visit www.nkgenbiotech.com .
Forward-Looking Statements
Statements contained in this press release may contain “forward-looking statements” within the meaning of Section 27A of the Securities Act and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements may be identified by the use of words such as “anticipate”, “believe”, “could”, “continue”, “expect”, “estimate”, “may”, “plan”, “outlook”, “future” and “project” and other similar expressions that predict or indicate future events or trends or that are not statements of historical matters. Because such statements are subject to risks and uncertainties, many of which are outside of the Company’s control, actual results may differ materially from those expressed or implied by such forward-looking statements. Such statements include, but are not limited to, statements regarding the Company’s plans and expected timing for developing SNK01, including the expected timing of completing and announcing further results from its ongoing clinical studies; and the Company’s expected timing for developing its product candidates and potential benefits of its product candidates. Risks that contribute to the uncertain nature of the forward-looking statements include: the Company’s ability to execute its plans and strategies; risks related to performing clinical studies; the risk that initial and interim results of a clinical study do not necessarily predict final results and that one or more of the clinical outcomes may materially change as patient enrollment continues, following more comprehensive reviews of the data, and as more patient data become available; potential delays in the commencement, enrollment and completion of clinical studies and the reporting of data therefrom; the risk that studies will not be completed as planned; the risk that the abstract will not be published as planned including delays in timing, format, or accessibility; and NKGen’s ability to raise additional funding to complete the development of its product candidates. These and other risks and uncertainties are described more fully under the caption “Risk Factors” and elsewhere in the Company’s filings and reports, which may be accessed for free by visiting the Securities and Exchange Commission’s website at www.sec.gov and on the Company’s website under the subheading “Investors—Financial and Filings”. Investors should take such risks into account and should not rely on forward-looking statements when making investment decisions. All forward-looking statements contained in this press release speak only as of the date on which they were made. The Company undertakes no obligation to update such statements to reflect events that occur or circumstances that exist after the date on which they were made, except as required by law.
Internal Contact: Denise Chua, MBA, CLS, MT (ASCP) Vice President, Investor Relations and Corporate Communications 949-396-6830 [email protected]
External Contacts: Chris Calabrese Managing Director LifeSci Advisors, LLC [email protected]
Kevin Gardner Managing Director LifeSci Advisors, LLC [email protected]
IMAGES
VIDEO
COMMENTS
Age usually >65 in sporadic cases (Late Onset AD). 11% of older persons may have Alzheimer's Disease. 60-70% of persons living in nursing facilities may have Alzheimer's. F>M. Mean duration ~8 yrs. Range 2-20 yrs. Early Onset AD in 50s is extremely rare. Genetic mutations in Amyloid Precursor Peptide.
Alzheimer's disease is the most common cause of dementia — a gradual decline in memory, thinking, behavior and social skills. These changes affect a person's ability to function. About 6.5 million people in the United States age 65 and older live with Alzheimer's disease. Among them, more than 70% are 75 years old and older.
Alzheimer's disease is a brain disorder that slowly destroys memory and thinking skills, and eventually, the ability to carry out the simplest tasks. In most people with Alzheimer's, symptoms first appear later in life. Estimates vary, but experts suggest that more than 6 million Americans, most of them age 65 or older, may have Alzheimer ...
History. Patients with Alzheimer disease (AD) most commonly present with insidiously progressive memory loss, to which other spheres of cognition are impaired over several years. This loss may be associated with slowly progressive behavioral changes. After memory loss occurs, patients may also experience language disorders (eg, anomic aphasia ...
Dementia is a general term that refers to a decline in cognitive ability severe enough to interfere with activities of daily living. Alzheimer's disease (AD) is the most common type of dementia, accounting for at least two-thirds of cases of dementia in people age 65 and older. Alzheimer's disease is a neurodegenerative disease with insidious onset and progressive impairment of behavioral and ...
This presentation entitled, Alzheimer's Disease - What is the Role of Public Health?, is part of a curriculum for public healt\൨ students entitled, A Public Health Approach to Alzheimer's and Other Dementias. It was developed by the Emory Centers for Trai對ning and Technical Assistance for the Alzheimer's Association with funding ...
Alzheimer's disease is a brain disease that slowly destroys memory and thinking skills. It is a progressive disease, which means it gets worse over time. Alzheimer's is the most common cause of dementia among people aged 65 and older. Rarely, people younger than 65 can have Alzheimer's. This is called early-onset Alzheimer's disease. This slide show explains the symptoms, causes and risk ...
Alzheimer's disease is the most common cause of dementia, accounting for at least two-thirds of dementia cases in people 65 and older. Other common causes of dementia include: Vascular dementia. Dementia with Lewy bodies. Frontotemporal dementia. Dementia due to Parkinson's disease. Advertisement.
Alzheimer's disease is a brain disorder that slowly destroys memory and thinking skills and, eventually, the ability to carry out the simplest tasks. In most people with the disease — those with the late-onset type symptoms first appear in their mid-60s. Early-onset Alzheimer's occurs between a person's 30s and mid-60s and is very rare.
1. Introduction. Alzheimer's disease (AD) (named after the German psychiatric Alois Alzheimer) is the most common type of dementia and can be defined as a slowly progressive neurodegenerative disease characterized by neuritic plaques and neurofibrillary tangles (Figure 1) as a result of amyloid-beta peptide's (Aβ) accumulation in the most affected area of the brain, the medial temporal ...
Alzheimer disease (AD) is a neurodegenerative disorder of uncertain cause and pathogenesis that primarily affects older adults and is the most common cause of dementia [ 1 ]. The most essential and often earliest clinical manifestation of AD is selective memory impairment, although there are exceptions. While treatments are available that can ...
Alzheimer disease is a neurodegenerative disorder that causes cognitive impairment. This Primer by Knopman et al. reviews the epidemiology of cognitive manifestations and risk factors, summarizes ...
ALZ Talks Virtual Events. Our free ALZ Talks webinars provide education, information, news and resources on a variety of dementia and caregiving topics. Register for an ALZ Talks webinar — brought to you by the Alzheimer's Association, with support from Procter & Gamble.
An important part of diagnosing Alzheimer's disease includes being able to explain your symptoms. Input from a close family member or friend about your symptoms and their impact on your daily life helps. Tests of memory and thinking skills also help diagnose Alzheimer's disease. Blood and imaging tests can rule out other potential causes of the ...
Signs & Symptoms: • Memory loss for recent events • Progresses into dementia almost total memory loss • Inability to converse, loss of language ability • Affective/personality disturbance • Death from opportunistic infections, etc. 7. Alzheimer's Disease: Postmortem Analysis normal advanced Alzheimer's. 8.
Soujanya Pharm.D. Definition, epidemiology, etiology, risk factors, pathophysiology, diagnosis and management of Alzheimer's disease. Health & Medicine. 1 of 24. Download now. Download to read offline. Alzheimer's Disease: Pathophysiology, Symptoms and Management - Download as a PDF or view online for free.
Free Google Slides theme and PowerPoint template. Raise awareness of Alzheimer's Disease with this template. Featuring a powerful color palette and a modern design, you can use this to reach out to your audience and educate them on the effects of this debilitating condition. Use the infographics to showcase facts and statistics, and use the ...
PK !ÀÚ§/i NA [Content_Types].xml ¢ ( ÌœÛrÚ0 @ß;Ó ðøµ Æ—&i ÈC/O½d&é ¨¶·¶¤±D þ¾² ‰É @²«Y½0Èö®Ž |Ö—…éå]] ·¼Ñ¥ ³0 O€‹\ ¥XÎÂ_7_G a «¤à³pÃux9 ûfz³Q\ 6ZèY¸2F}Œ" ¯xÍôX*.ìš…ljfì°YFŠå Ù'GÉdr åR .ÌÈ´9Âùô3_°ue‚/wvqO¢Ä2 >õÛµSͲnãÛåÑÁˆ?Š éV Žix¥ Å0¥ª2gÆ® nEñh_FÛý ÛÈn ½*•~g7xb†vÍÓ ...
Alzheimer's disease ppt. One of the hallmarks of Alzheimer's disease is the accumulation of amyloid plaques between nerve cells (neurons) in the brain. Amyloid is a general term for protein fragments that the body produces normally. Beta amyloid is a protein fragment snipped from an amyloid precursor protein (APP).
Alzheimer's disease (AD) is characterized by two pathological lesions, beta-amyloid (Aβ) plaques and neurofibrillary tangles, that are progressively distributed throughout the brain. These lesions are associated with disruption of the communication pathways between neurons, neuron degeneration, brain atrophy, and functional loss.
Inform and move your audience with these Alzheimer's disease awareness slides, easy to use as a Google Slides template, PowerPoint theme or Canva template. Fill out professionally-designed, ready-made pages for symptoms and diagnosis, treatment options, statistics, and the latest research. Choose your own color scheme and font combination ...
Uncategorized. Alzheimer's disease is a progressive neurological condition. Common symptoms include memory loss, language difficulties, and more.
Alzheimer's Breakthrough Presentation . Medical . Free Google Slides theme and PowerPoint template . After doing research on Alzheimer, you have just discovered something incredible. Share your results with this simple template. Use our illustrations, graphic resources and infographics to talk about methodologies, treatments and any other detail.
Alzheimer's disease (AD) is the predominant dementia globally, with heterogeneous presentation and penetrance of clinical symptoms, variable presence of mixed pathologies, potential disease subtypes, and numerous associated endophenotypes. Beyond the difficulty of designing treatments that address the core pathological characteristics of the ...
Presentation: "Interrogating N-linked protein glycosylatin metabolium in mouse and human models of Alzheimer's disease" Baylea Davenport. Concentration: Genetics. Mentor: Helen Jones, Ph.D. Presentation: "Mitigation of placental inefficiency and fetal growth restriction through IGF1 gene therapy in a guinea pig model"
Alzheimer's disease causes significant problems with memory, thinking and behavior and is the most common form of dementia, affecting more than 50 million people around the world each year.
The interactive event kicked off with an introductory presentation on Alzheimer's disease and brain health from Dr. Karen Torres, a bi-cultural, bi-lingual neuropsychologist from the ADRC. Afterwards, attendees cycled through interactive learning stations where they could learn about brain health, nutrition, and physical activity.
There's still time to download this template and create a presentation in honor of World Alzheimer's Day, which is celebrated on September 21. Since it's a disease that has no cure yet, we have used a design that eases the atmosphere and softens the mood. For example, we've included rounded shapes and very light tones of purple. There are also ...
Song will also present clinical safety and efficacy data from the Company's recently completed Phase 1 clinical trial evaluating autologous SNK01 NK cell therapy in patients with Alzheimer's ...
NKGen Biotech Announces Upcoming Presentation on SNK01 NK Cell Therapy in Neurodegenerative Disease at the 12th Annual Alzheimer's & Parkinson's Drug Development Summit. Published: Apr 18, 2024. ... in Alzheimer's disease at the 12 th Annual Alzheimer's & Parkinson's Drug Development Summit to be held in Boston, MA, from April 23-25